

Assembler un génome est une tâche fastidieuse et surtout très coûteuse. Pour un génome de 100 Mbp (Million de paires de bases), Velvet ou SOAPdenovo utilisent 20 à 30 Go de mémoire, voire plus. Je voudrais partager avec vous une idée complètement farfelue qui nous est venue et qui nous a fait gagner (Guillaume Rizk et moi-même) le prix du meilleur poster lors de JOBIM 2013 : et si on faisait de l'assemblage de génome sur un Raspberry Pi ?

Remettons-nous d'abord dans le contexte…
L'assemblage de génome
Je ne vais pas décrire ici tout ce qui concerne l'assemblage de génome, cela méritera un article entier… même plusieurs. Je vais simplement vous présenter les idées générales et surtout le cas qui nous concerne : l'assemblage de novo.
L'assemblage de génome consiste à reconstituer un génome à partir d'un ensemble de reads, des séquences d'ADN "lues". Ces reads peuvent provenir de divers types de séquenceurs. Nous en avons présenté quelques uns dans l'article : Le séquençage, une histoire de génération. En fonction des technologies, les reads peuvent être plus ou moins longs et produits plus ou moins rapidement.
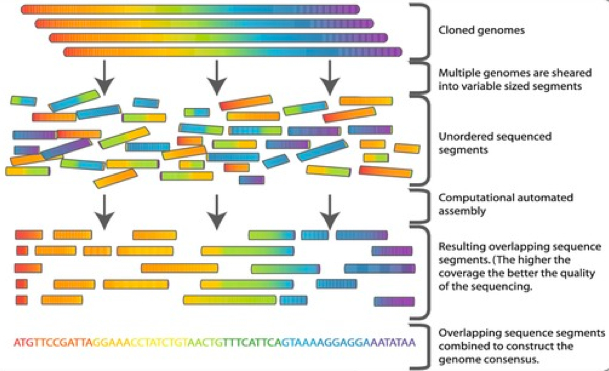
Dans notre cas, nous avons des reads petits (~100 bp) mais en grande quantité et nous n'avons pas de génome de référence. La procédure largement utilisée dans ce cas là se compose, en gros, de trois étapes :
- Le comptage des k‑mers
- L'assemblage des contigs
- La création de scaffolds
Pour faire simple, les reads sont découpés en morceaux chevauchants de k caractères : les k‑mers. Les k‑mers sont comptés afin de supprimer ceux qui ne sont pas suffisamment représentés et qui sont considérés comme des erreurs de séquençage.
À partir des k‑mers chevauchants, nous pouvons ensuite reconstituer des séquences d'ADN contiguës, les contigs. Cette étape est généralement réalisée en utilisant un graphe de De Bruijn. Enfin, puisqu'il reste des trous entre les contigs, dans une dernière étape, ils seront rassemblés en scaffolds (échafaudages) dans lesquels la taille des trous pourra être estimée.
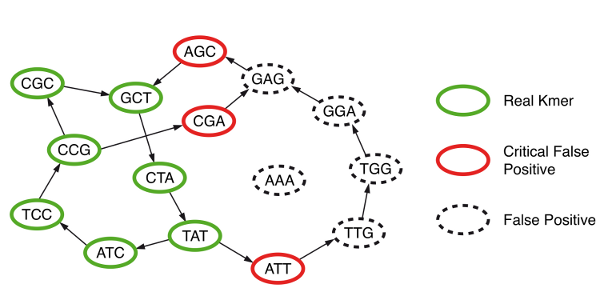
k‑mers faux positifs (noeuds rouges et noirs pointillés) (CC BY-NC-ND 3.0)
Les deux premières tâches sont celles qui nous intéressent particulièrement car elles sont les plus difficiles. En effet, elles demandent de stocker tous les k‑mers en mémoire. Or, si nous voulons compter des 30-mers, il nous faut une table qui contienne au pire 430 entiers… même si chacun d'eux était encodé par un seul octet, ça fait tout de même 109 Go… ça monte vite non ? On n'arrive jamais à cette extrémité mais cela reste très gourmand.
MINIA, un assembleur minimaliste
Afin de réduire la quantité de mémoire vive utilisée pour l'assemblage, Rayan Chikhi, Guillaume Rizk et Dominique Lavenier, ont eu l'idée d'utiliser une structure de données informatique particulièrement compacte, le filtre de Bloom, que Nico vous a déjà présenté ici. Je ne reviens donc pas sur la définition.
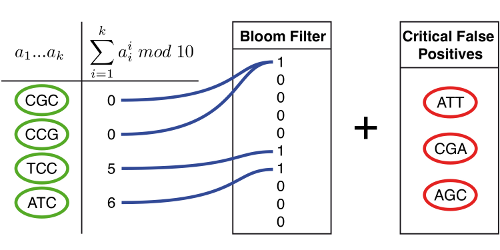
Le graphe de De Bruijn est encodé via un filtre de Bloom qui nous dit si un k‑mer y est présent ou non. Mais pour ne pas nous tromper, il faut que le filtre soit parfait, or nous savons qu'il ne l'est pas. Ce problème est résolu en gardant, dans une autre table, les k‑mers non-présents mais qui sont voisins d'un k‑mer présent dans le graphe (cercles rouges). Ainsi, nous sommes sûrs de ne jamais nous tromper.
Cela résout le problème de la taille du graphe de De Bruijn mais pas celui du comptage des k‑mer. Pour ce problème, la réflexion a été la suivante : "puisque nous n'avons pas assez de mémoire vive, pourquoi ne pas utiliser le disque dur ?" Ainsi, une procédure par découpage de l'espace des k‑mers a été utilisée comme illustrée dans l'image ci-dessous. Chaque read est lu et découpé en k‑mers. Puis une fonction de hachage est appliquée à chaque k‑mer afin de savoir à quelle partition il appartient. Chaque partition est écrite sur le disque dur. Une fois que tout est lu, chaque partition est chargée en mémoire, triée, comptée puis réécrite sur le disque. Évidemment, la taille des partitions est calculée de façon à optimiser l'utilisation de la mémoire vive.
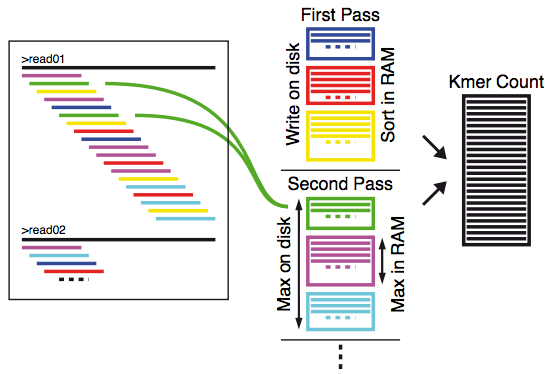
Ces deux idées ont été implémentées dans le logiciel MINIA et sont plus largement décrites dans les références suivantes :
- R. Chikhi, G. Rizk. Space-efficient and exact de Bruijn graph representation based on a Bloom filter, WABI 2012
- G. Rizk, D. Lavenier, R. Chikhi. DSK : k‑mer counting with very low memory usage, Bioinformatics, 2013.
Mettons tout ça sur un Raspberry Pi
Là ! Y a défi ! Même si MINIA est peu gourmand en ressources, ça reste difficile de faire tourner un assembleur avec seulement 512 Mo de RAM. Car notre Raspberry Pi est vraiment un tout petit système informatique : un processeur ARM à 700MHz, 512 Mo de RAM, une carte SD de 4 Go pour le GNU/Linux et une clé USB de 32 Go pour les données. C'est pas énorme !
D'ailleurs, lors des premiers essais sur E. coli, cela ne fonctionnait pas. Les buffers d'écriture sur fichiers étaient trop grand (2 Mo par fichier x 100 fichiers ouverts = 200 Mo). Après correction, nous avons réussi à assembler le génome de E. coli puis celui de C. elegans. Ça devient intéressant lorsqu'on compare à Velvet et SOAPdenovo qui utilisent respectivement 30.6 et 29.6 Go de RAM pour assembler C. elegans, là où MINIA en utilise moins de 300 Mo.
Par contre, c'est vrai que le Raspberry Pi est lent, il lui a fallu 18h pour réaliser l'assemblage. Ceci dit, sur un ordinateur de bureau, Velvet a mis 13h… MINIA, sur une machine largement moins performante, s'en tire plutôt pas mal. Bref, tous ces résultats sont donnés sur notre poster disponible ici ou là.

Enfin, pour JOBIM, nous avions besoin de faire la démonstration en direct. Guillaume Rizk et moi-même avons donc sorti le fer à souder, deux trois LED, une mini imprimante thermique, un écran LCD 2x16 caractères, des câbles, etc… Et nous avons créé le Picontigotron : un assembleur de génome dans une boite à chaussures ! La classe totale. 🙂

Conclusion
Le but de ce billet, au-delà du plaisir de vous raconter ma petite histoire, n'est pas de vous dire que MINIA est le meilleur assembleur de tous les temps, loin de là. D'ailleurs, il ne fait que les deux premières étapes de l'assemblage, il ne sait pas construire les scaffolds. Ce n'est pas non plus de vous inciter à acheter des Raspberry Pi…
Non, ce que j'espère vous faire passer, c'est que désormais, il va falloir être malin. Aligner les ordis dans des clusters n'est pas forcément LA solution. Avec des structures de données bien pensées et adaptées aux problèmes, avec des algorithmes qui utilisent finement les ressources des ordinateurs, nous pouvons réaliser des tâches extrêmement complexes sur un ordinateur de bureau. Nous avons bien réussi sur un Raspberry Pi.
La bioinformatique est en cela une source merveilleuse de problèmes pour les informaticiens. J'adore bosser dans ce domaine, j'espère que vous aussi. 😉
Je remercie Nolwenn, Max et Sylvain pour leur relecture.
Laisser un commentaire