

Étiquette : réseau métabolique
-
Représenter le métabolisme - les réseaux métaboliques
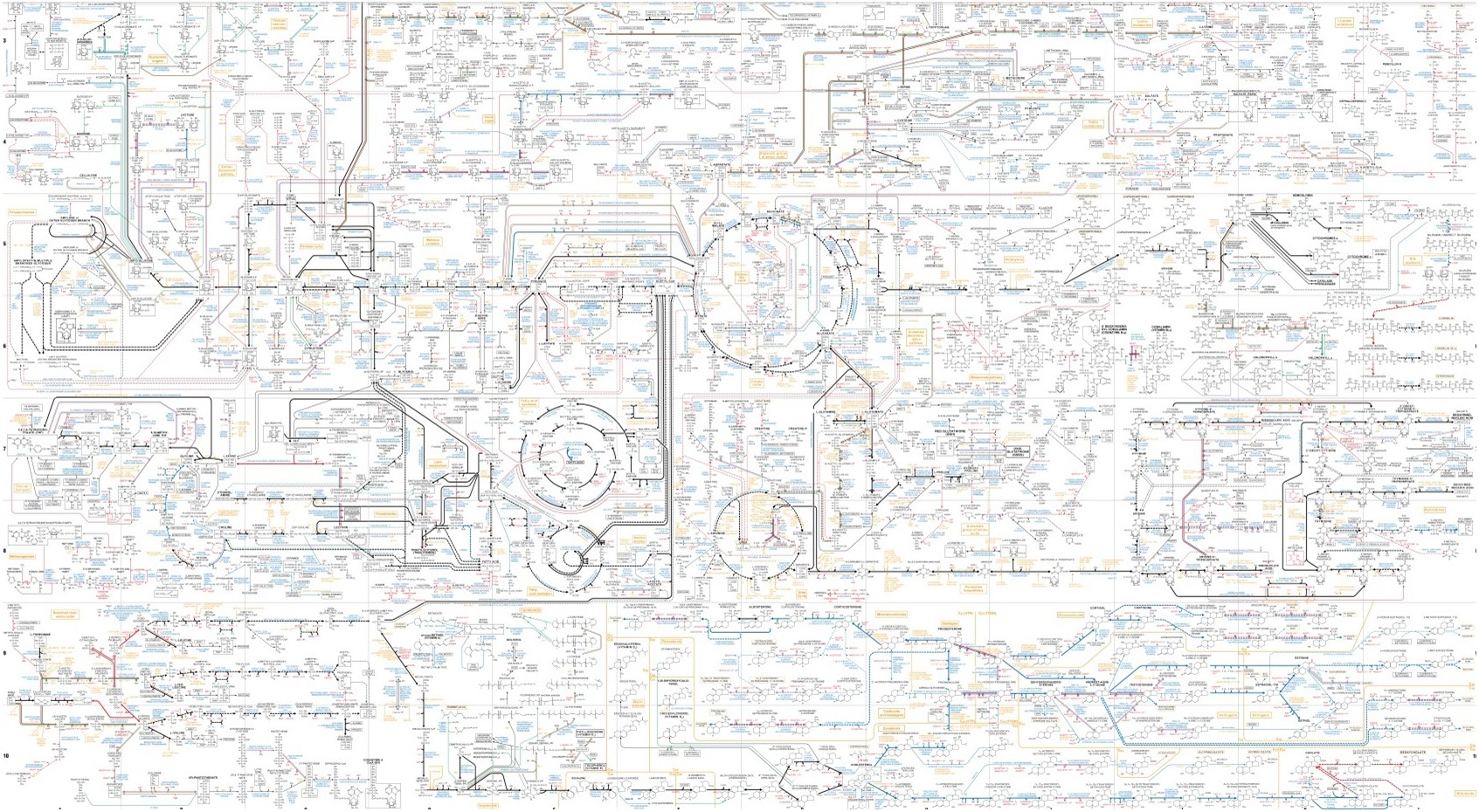
*grosse voix très sérieuse* Attention ! L'article qui suit est le premier d'une série d'articles sur la représentation du métabolisme sous forme de réseaux et leur analyse. Il existe, en bioinformatique, plusieurs catégories de modèles pour décrire le métabolisme.Tout d’abord, les modèles pour l’analyse structurelle du métabolisme. Cette catégorie regroupe principalement les modèles reposant sur la théorie…
-
Flux Balance Analysis ou la simulation du métabolisme d'une cellule
"I thought there couldn't be anything as complicated as the universe, until I started reading about the cell.“ (Systems Biologist Eric de Silva -astrophysicist by training-, Imperial College London.) Avec l'arrivée des technologies de séquençage à haut débit (NGS pour les intimes), de plus en plus de génomes complets sont disponibles dans les banques de données…
-
Les identifiants : a (name)space oddity

Sur Bioinfo-fr.net, nous avons plusieurs fois parlé des bases de données biologiques. Que ce soit du point de vue de la gestion, de l'exploitation ou même du stockage physique. J'aimerai revenir aujourd'hui sur un souci qui se présente souvent lors de l'exploitation de plusieurs bases de données : les identifiants. Un objet biologique dans une base…
-
La complétion de réseaux métaboliques
Il y a quelques temps, sur ce blog, j'ai publié un article qui parlait de la reconstruction automatique de réseaux métaboliques à partir d'un génome annoté. Dans cet article je vous ai promis de continuer à parler de ce sujet, notamment pour voir comment l'on pouvait améliorer la qualité d'un réseau métabolique fraichement créé. Pour…
-
Reconstruction automatique de réseaux métaboliques

Le nombre de génomes séquencés croît aujourd'hui exponentiellement. Il est probable que d'ici peu de temps chacun d'entre nous puisse avoir la séquence de son propre génome pour une poignée de dollars en quelques jours seulement. On peut d'ailleurs se référer à ce récent débat vidéo, dans lequel intervient notamment le professeur Denis Duboule (généticien…