

Étiquette : NCBI
-
Les bases de données de séquençage : GEO, SRA, ENA, ArrayExpress
De nos jours, lors de la publication de résultats, il est nécessaire de rendre public les éventuelles données de séquençage générées. Si un faible nombre d’irréductibles continuent à ne fournir les données que sur demande, les bonnes pratiques poussent à les déposer dans des bases de données librement accessibles. Quatre grandes bases de données de séquençage…
-
Le site du NCBI "un peu" plus moderne grâce aux extensions de navigateurs

« Pour la semaine prochaine vous allez devoir trouver et étudier l’article sur le site du NCBI [insérer un nom bien compliqué ici] » Passée la déception d’avoir à se plonger dans un article obscur en anglais, on ouvre l’article en question et là… on découvre un énorme pavé de texte ma foi fort intéressant mais…
-
L'analyse de données RNA-seq : mode d'emploi

Un jour, un biologiste se pointe chez vous avec d'une part un disque dur externe dans la main, d'autre part l'air soucieux. Il veut que vous analysiez ses données RNA-seq. Le disque, c'est parce qu'il a environ 50Gb de données à vous transmettre ; l'air soucieux, c'est parce qu'elles ont coûté dans les 15'000 euros, et…
-
Chercher des motifs dans un fichier
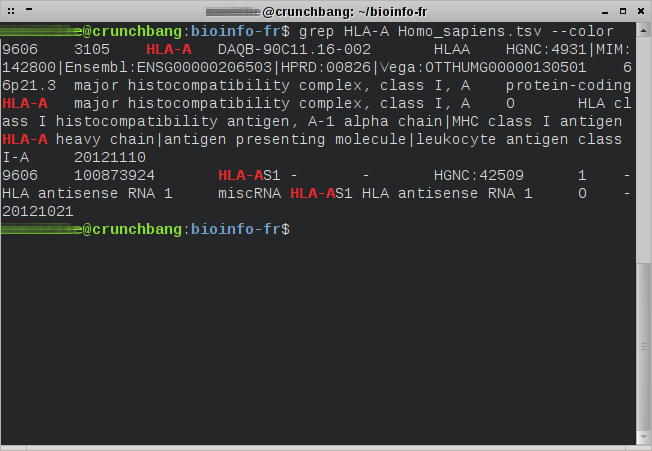
Langage : shellCommandes présentées : grep, split (succintement)Niveau : débutant Présentation de la commande grep La commande grep est disponible nativement sur la plupart des systèmes d'exploitation GNU/Linux. La plupart des utilisateurs utilisent cette commande pour rechercher un mot ou un groupe de mots, que nous appellerons motif (pattern en anglais), dans un fichier texte. Cependant cette commande…
-
Astuce programmation BioPython : Parser les multi-genbank et les multi-FASTA produits par Batch Entrez
Prérequis : Savoir 'un peu' se servir d'un shell et avoir installé Python et son module Bio. But : Redécouper des multi-genbank ou des multi-FASTA en un fichier par entrée. Difficulté : 2/5 (Facile) Principe : Le NCBI propose un outil très pratique pour récupérer facilement des jeux de données diversifiés : BatchEntrez, vous trouverez plus d'information ici. On télécharge ainsi un…
-
Récupérez facilement des données hébergées par le NCBI : BatchEntrez
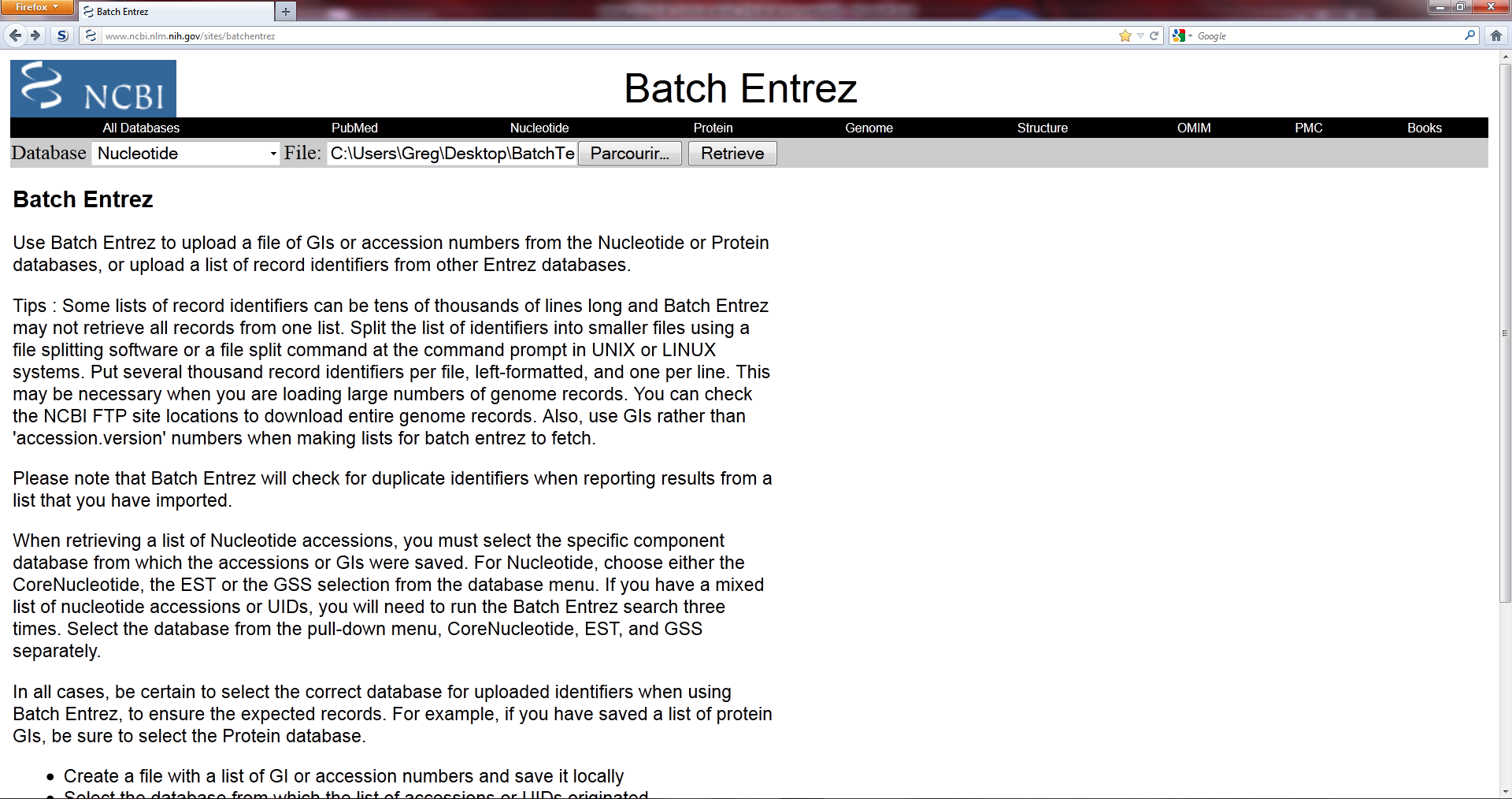
But : Les bases de données du NCBI abritent de très nombreuses informations : génomes, protéines, références bibliographiques, etc. Si vous souhaitez récupérer l'une d'entre-elles, une recherche sur le site est la solution la plus simple, mais si vous avez besoin de récupérer de nombreuses données dans un des formats proposés, alors le NCBI a mis l'outil…
-
Récupérer la fiche d'un gène avec les Eutils du NCBI
En bioinformatique il n'est pas rare que l'on ait besoin d’accéder à des informations disponibles sur des bases de données internationales, nous verrons ici le cas de la banque Gene du NCBI. Avant de s'intéresser à la récupération d'une fiche d'un gène en passant par les Eutils, un peu de théorie et d'explications sur une fiche…