

Catégorie : Découverte
-
IA et analyse des génomes : un autre révolution en cours ?(partie 3)
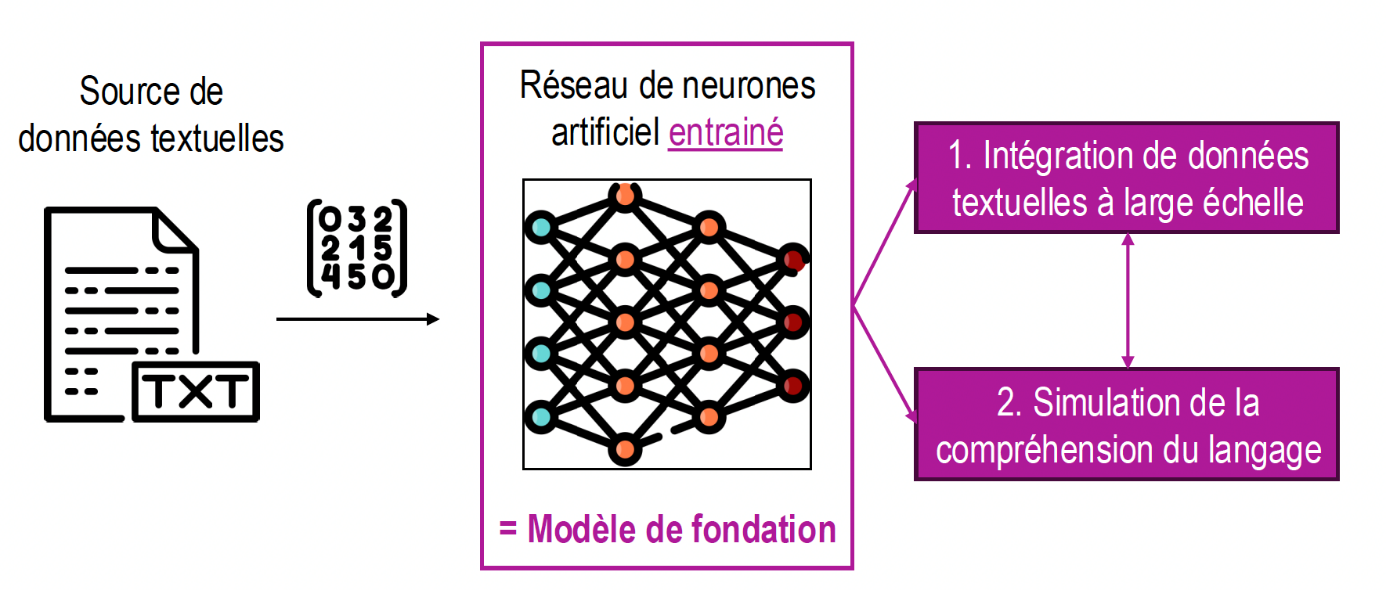
Dans cette dernière partie (et bravo si vous avez lu les deux premières !), nous avons essayé de prendre un peu de recul sur les conséquences de la disponibilité de modèles de fondation tels que Evo2. En particulier, la question des usages à venir des bases de données de séquences nous semble importante. Egalement, la…
-
IA et analyse des génomes : une autre révolution en cours ? (partie 2)
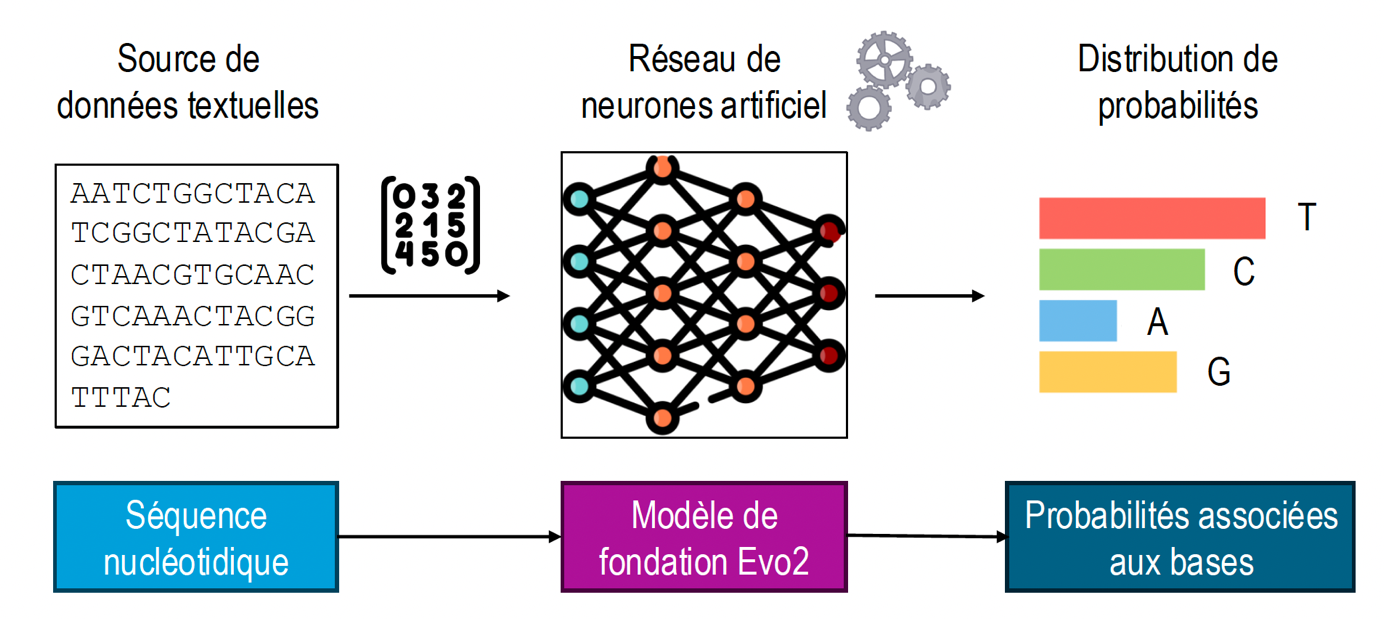
Si vous avez lu la partie 1 de cette série de trois articles, vous aurez compris que l'étude de l'article de présentation du modèle Evo2 (Brixi et al., 2025) nous aura littéralement enflammées au laboratoire 🤯. Le texte est à la fois dense et exigeant, mais il offre une contribution véritablement nouvelle au domaine. Ci-dessous,…
-
IA et analyse des génomes : une autre révolution en cours ? (partie 1)
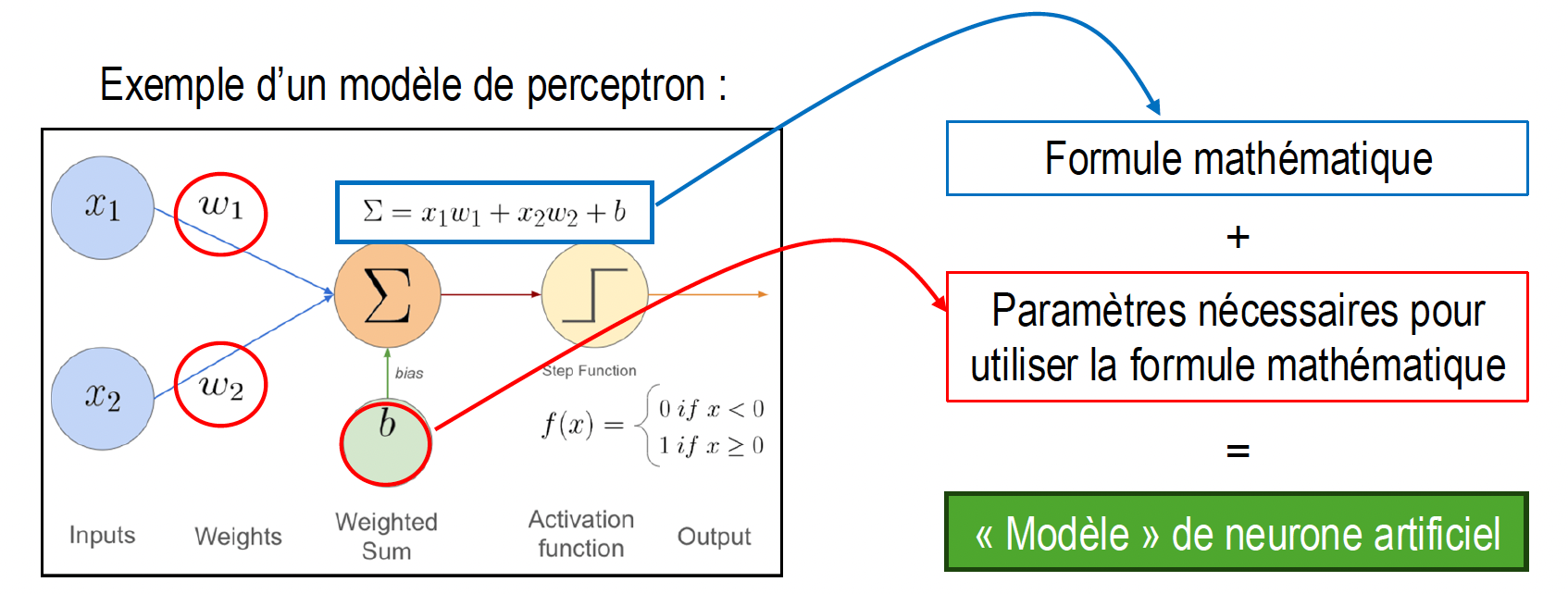
Cher·es toutes et tous ! À chaque fois que je parcours ce blog, je me dis : « Ouah, c'est génial de prendre le temps, ainsi, de partager ses idées et ses savoir-faire, mais pourquoi est-ce que je ne le fais pas plus souvent ? ». Ma dernière (et unique !) contribution date en effet de 2021. Cela fait…
-
La fin du génome linéaire : Bienvenue dans l'ère (complexe) du Pangénome
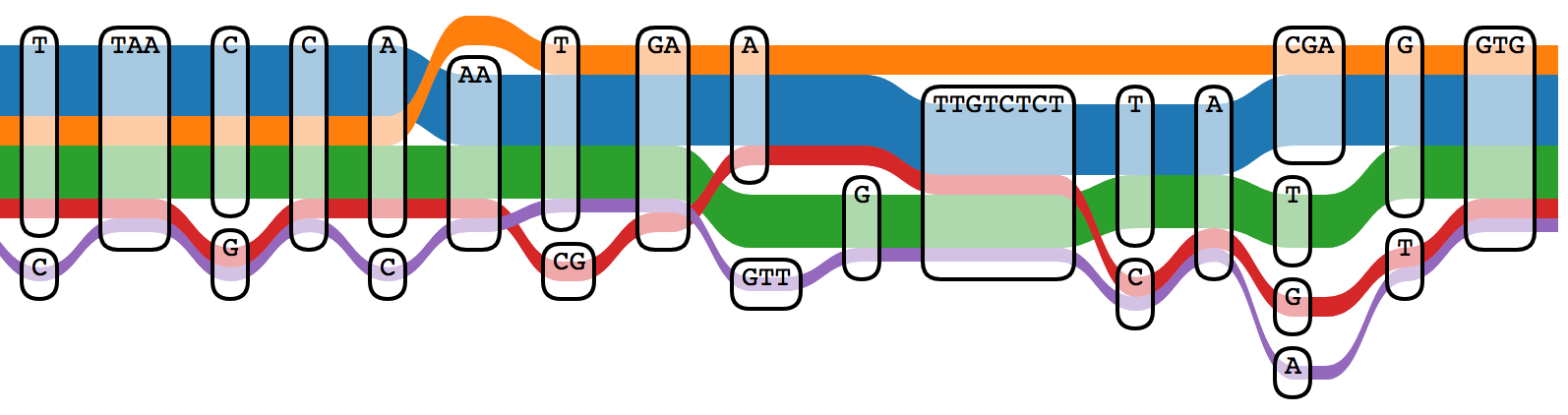
Que vous soyez bio-informaticien aguerri, étudiant en master ou simplement curieux ayant déjà manipulé un fichier FASTA un jour dans votre vie, ce billet de blog est pour vous. Nous allons explorer pourquoi et comment nos repères actuels en génomique sont en train de changer de forme. Le génome de référence classique Prenons l'exemple du…
-
La complexité inattendue des systèmes de coordonnées en bioinformatique
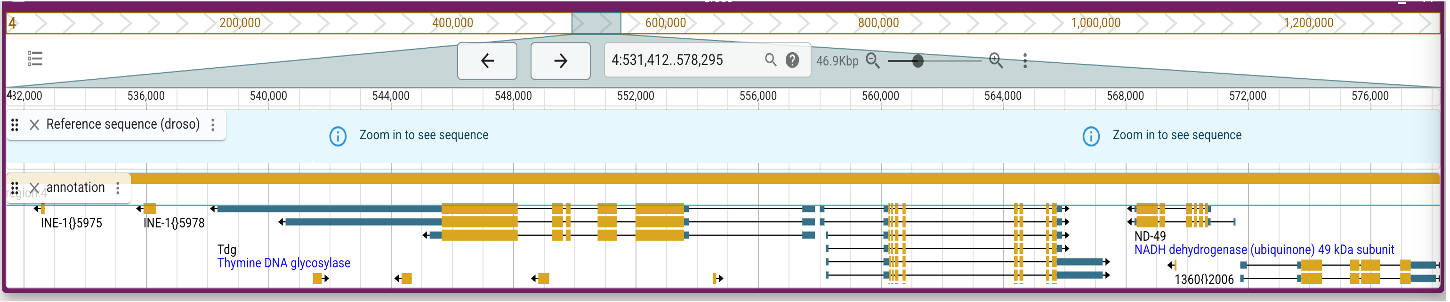
En bioinformatique, quand on manipule des génomes, des annotations, des fichiers d’alignement, des variants ou des intervalles (exons, régions régulatrices, etc.), on est rapidement confronté à deux notions fondamentales qui conditionnent la manière dont les positions sont décrites et interprétées : Ces deux dimensions définissent ensemble le système de coordonnées utilisé. Les maîtriser permet d’interpréter correctement…
-
Trois exemples concrets qui m'ont fait m'intéresser à la reproductibilité entre langages
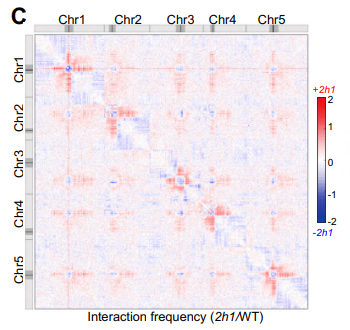
Mes très chers confrères adorés, Comme vous le savez si vous lisez mes excellents articles d'opinion, tous géniaux (hum !), cela fait quelques années maintenant que je travaille pour des toxicologues. Alors, c'est bien, j'ai appris plein de choses, au point de me définir aujourd'hui comme « computational toxicologist ». Mais j'ai surtout mis mon nez dans…
-
L'alignement multiple en étoile ⭐
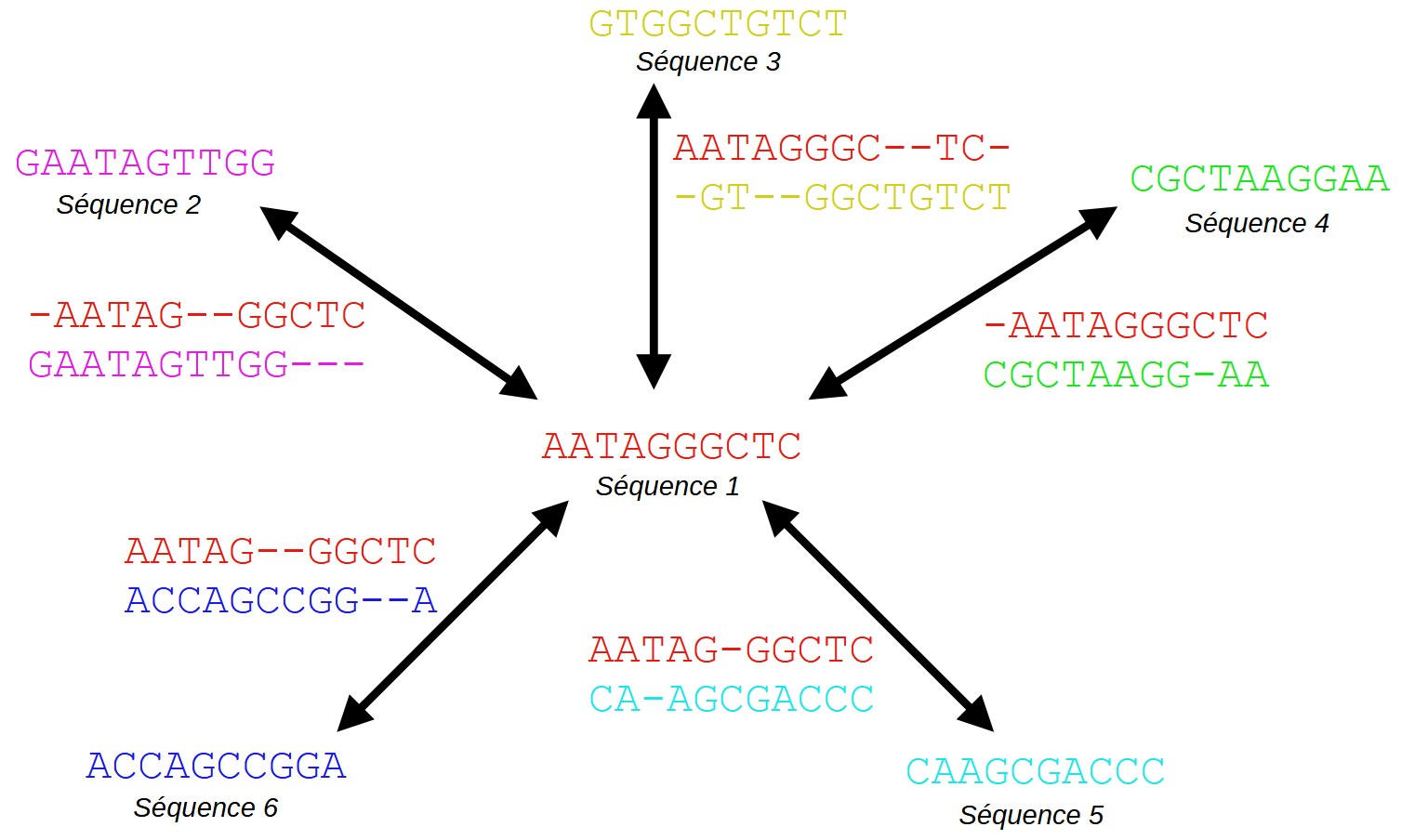
Introduction Dans le domaine de la bioinformatique l'alignement de séquences est une opération courante pour comparer des séquences biologiques (ADN, ARN, protéines). Nous avons vu dans un précédent article le principe de l'algorithme de Needleman-Wunsch qui produit un alignement global optimal de deux séquences. Au-delà de l'alignement par paire, l'alignement multiple de séquences (Multiple Sequence…
-
Alignement de séquences : bien comprendre l'incontournable algorithme de Needleman-Wunsch
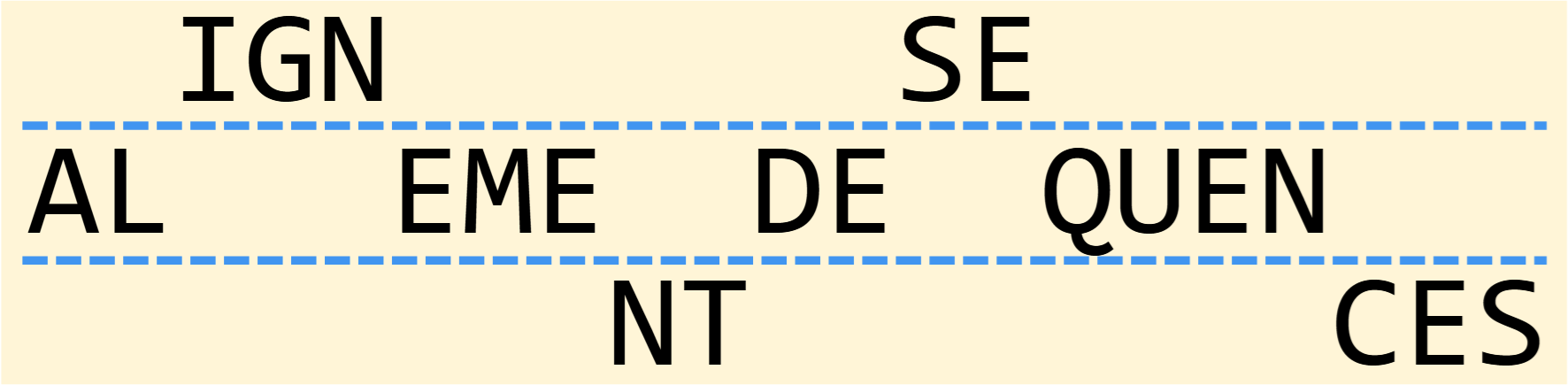
Introduction L'analyse des séquences biologiques (ADN, ARN ou protéines) est un sujet central de la biologie moléculaire et de la bioinformatique. Dans ce cadre, chercher à savoir à quel point deux séquences sont similaires ou dissimilaires est une des questions qu'on se pose le plus souvent (par exemple dans le but d'analyser des relations phylogénétiques).…