

Quand j'étais étudiante en master, la grande majorité des cours de structure étaient réservés aux protéines, ce qui fait que la structure des ARN n'a pas été très développée au cours de mon cursus. Du coup, je vous livre ici une partie de ce que j'ai appris en stage.
Définition 0 : ARN
L'ARN (acide ribonucléique) est une macromolécule biologique qui remplit de nombreuses fonctions biologiques telle que :
- support intermédiaire de la traduction de l'ADN en protéines,
- catalyseur de l'activité peptidyltransférase menant à la formation d'une liaison peptidique,
- régulateur de l'expression génique grâce aux riboswitch ou a de petites molécules interférenetes,
- ou encore l'inactivation de chromosome par de longs ARN interférents.
Si la conservation de la séquence est d'une importance primordiale pour le fonctionnnement d'une protéine, ce dogme central est remis en question dans le cadre des ARN. En effet, il semblerait que les facteurs important pour que les ARN puissent mener à bien leurs rôles soient la structure secondaire et la structure tertiaire des ARN.
Définition 1 : Structure d'un ARN
La structure 3D d'un ARN est la donnée des coordonnées de ses atomes dans l'espace, données que l'on peut retrouver dans un fichier de la PDB. À partir de ces données, on peut utiliser PyMOL[5] pour représenter la structure 3D de l'ARN considéré, comme sur la Figure 1

Dans cet article, nous allons nous intéresser en particulier à la structure secondaire des ARN, avec quelques apports de structures 3D afin de pimenter un peu l'histoire. L'objectif final est d'être prêt à affronter l'alignement de structures secondaire c'est-à-dire que vous puissiez utiliser du code qui génère les structures secondaires d'ARN à partir de fichiers PDB et, puisque nous y sommes, d'autres informations pertinentes sur les ARN.
Représentation des structures d'ARN
Avant toutes choses (y compris définir proprement ce qu'est une structure secondaire) intéressons-nous à la représentation des structures d'ARN : il en existe plusieurs qui ont leur intérêts et leurs limitations.
Très clairement, la représentation de la Figure 1 tirée des coordonnées atomiques directement est celle qui semble la plus « réaliste » mais elle n'est que peu appropriée pour la modélisation et la prédiction de structures.
La représentation en squiggle-plot, motifs d'intérêts et limitations
La représentation en squiggle-plot est celle qui se rapproche le plus de ce que l'on pourrait dessiner manuellement et « naturellement » pour représenter un ARN. C'est la représentation qui permet de « comprendre » le mieux ce qu'il se passe dans la structure comme on peut le voir sur les Figures 2 à 5. Ces figures représentent les motifs d'intérêts les plus communs dans les ARN et leur nomenclature.




En revanche, pour l'alignement de séquence ou de structure, elle est vraiment (vraiment) pas top, comme expliqué dans une review de la comparaison de structures secondaires de Schirmer et al [1].
En particulier, ce genre de dessin est produit par des algorithmes dont les objectifs principaux sont l'esthétique et l'évitement des recouvrements des sous-structures ce qui ne coïncide pas du tout avec la réalité.
La représentation en dot-parenthesis
La représentation en dot-parenthesis (ou séquence bien parenthésée ou encore dps) est celle que l'on peut voir Figure 6. Ce genre de représentation est utilisé comme entrée par le logiciel VARNA [2], qui permet de produire les images telles que celles présentées Figures 2 à 5. Ce genre de représentation est un bon compromis entre la lisibilité et la praticité.
Définition 2 : Représentation dot-parenthesis
Chaque structure de taille 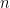
Chaque paire de bases appariées 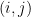
- le caractère à la
ème position est une parenthèse ouverte "("
- le caractère à la
ème position est une parenthèse fermante ")".
Chaque base non-appariée est représentée par un point ".".
Un exemple est donné en dessous.
|
1 2 3 |
00 01 02 03 04 05 06 07 08 09 10 G C A A U G C U U A A . . ( ( . . . ) ) . . |
Figure 6 : Représentation bien parenthésée (dps) d'une chaîne d'ARN.
Ce qu'il faut comprendre c'est que les bases
sont en interactions,
(03, 07)aussi.
La représentation en forme d'arbre
Si vous êtes informaticien, vous savez ce qui va venir… En fait, une séquence bien parenthésée c'est un arbre (oui je sais, j'ai l'air de faire de la magie mais je vous jure qu'il y a de vrais fondements à ce que je dis (promis je ne donnerais pas les détails)).
On va, pour une fois, commencer par donner un exemple plutôt que de donner une définition/algorithme.

Je suppose que vous voyez ce que ça fait ? Oui ? Non ? Pas convaincu ? Vous inquiétez pas, voici l'algorithme, écrit en C++ !
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 |
Tree< std::vector<int> >* dps::parse(int & index) { /* Point de départ de la récursion */ int start = index ; /* Si on est dans un appel récursif, il faut créer un arbre complet * qui deviendra un sous arbre quand retourné. */ Tree< std::vector<int> > * current_root = new Tree< std::vector<int> >(); /* Traversée, caractère par caractère de la séquence */ while (index < (int)this->sequence.length()) { index ++; switch (this->sequence[index - 1]) { case '.': { /* C'est une feuille */ std::vector<int> pos = { index - 1, index - 1 }; Tree< std::vector<int> >* leaf = new Tree< std::vector<int> >(pos); current_root->append_child(leaf); break ; } case '(': { /* Appel récursif pour trouver la fin de l'arbre */ Tree< std::vector<int> >* node = parse(index); current_root->append_child(node); break ; } case ')': { /* Tous les enfants sont découverts, on retourne le sous-arbre */ std::vector<int> current_pos = { start - 1, index - 1}; current_root->set_position(current_pos); return current_root ; break ; } default : { /* Ceci ne devrait pas arriver, mais bon… */ std::stringstream stream ; stream << "Sequence has an unexpected character that isn't ( . or )\n"; DPSInvalidInput myerror(stream.str()); throw myerror ; break ; } } } |
Algorithme 1 : Algorithme de transformation d'une séquence bien parenthésée en arbre
Bien évidemment, il ne fonctionne pas en stand alone, il faut la classe
Tree[T],la classe
dps(dot-parenthesis sequence) et la gestion d'erreur
DPSInvalidInputmais vous avez l'idée générale.
Le paramètre de la classe
Tree[T]est un vecteur d'entiers qui donne les positions dans la séquence correspondant au nœud interne ou à la feuille sur laquelle on travaille.
- Pour une feuille, l'étiquette est le couple {(int) position du ., (int) position du .}, c'est-à-dire la position du "." dans la séquence bien parenthésée.
- Pour un nœud interne, l'étiquette est le couple {(int) position de (, (int) position de ) correspondante}.
On remarquera que l'arbre que j'ai donné Figure 7 contient une racine
root (-1, -1)qui est racine « virtuelle », c'est-à-dire qu'elle ne correspond à aucune réalité biologique ou de la séquence.
C'est parce qu'en fait, là, dans la séquence
..((...))..le premier caractère est un "." et donc une feuille, ce qui fait qu'on obtient une foret (une « famille » d'arbre) si on ne prend pas garde d'ajouter cette racine « virtuelle ».
Il faut traiter ce cas à part dans le code, selon ce que l'on souhaite faire par la suite (dans le doute, ajouter systématiquement une racine virtuelle à pour être certain d'obtenir un arbre et pas une foret semble être une bonne initiative).
Qu'est-ce qu'une structure secondaire d'ARN ?
Définitions
La structure d'un ARN est le résultat d'appariemment de bases (oui oui, je sais que vous savez) grâce à des liaisons hydrogènes, nous appellerons ces liaisons des arcs et la donnée d'une structure avec ces liaisons une structure arc-annotée, comme sur la Figure 9.
Définition 3 : Arcs emboîtés et arcs indépendants
Soient 2 arcs 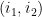
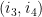
Ils sont dit indépendants s'ils respectent l'une des inégalités suivantes :
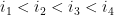

Ils sont dit emboîtés s'il respectent l'une des inégalités suivantes :
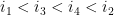
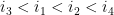
On a alors toutes les définitions (informatique) nécessaires pour définir la structure secondaire d'un ARN.
Définition 4 : Structure secondaire d'un ARN
La structure secondaire d'un ARN est le plus gros sous-ensemble d'arcs emboîtés et indépendants qui peuvent être extrait d'une séquence d'ARN arc-annotée.
Planarisation d'une structure secondaire d'ARN
Tout ceci est bien beau mais en vrai, dans un ARN il y a plein d'interactions contradictoires me direz-vous. Et vous avez parfaitement raison, on va devoir supprimer des arcs de la représentation arc-annoté, dont un exemple est disponible Figure 9, pour obtenir une séquence bien parenthésée comme celle de la Figure 6.
« Retirer » des arcs, c'est-à-dire des interactions, est un problème que l'on appelle planarisation de l'ARN, et qui nécessite l'introduction d'une définition avant d'être défini : la notion de structure générique.
Définition 5 : Structure générique
Une structure générique est un graphe 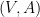
![V = [| 1; n |]](https://s0.wp.com/latex.php?latex=V+%3D+%5B%7C+1%3B+n+%7C%5D&bg=ffffff&fg=000&s=0&c=20201002)
![A \subseteq \{ (i, j) \in [| 1; n |]^2 | i < j \}](https://s0.wp.com/latex.php?latex=+A+%5Csubseteq%C2%A0%5C%7B+%28i%2C+j%29+%C2%A0%5Cin+%5B%7C+1%3B+n+%7C%5D%5E2+%C2%A0%7C+i+%3C+j+%5C%7D&bg=ffffff&fg=000&s=0&c=20201002)
Une structure générique 
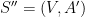

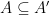
Nous voici équipés pour définir le problème de planarisation de l'ARN !
Problème 1 : planarisation d'une structure secondaire d'ARN
À partir d'une structure générique 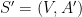
- inclusivité
- planarité
- optimalité
tels que
planaire,

![\forall (i, j) \neq (i', j') \in A', (i \neq i') \wedge (j \neq j') \wedge \left( [| i; j |] \cap [| i', j' |] = \left\{ \begin{array}{l} \emptyset \\ [| i; j |] \\ [| i'; j' |] \end{array} \right. \right)](https://s0.wp.com/latex.php?latex=+%5Cforall+%28i%2C+j%29+%5Cneq+%28i%27%2C+j%27%29+%5Cin+A%27%2C+%28i+%5Cneq+i%27%29+%5Cwedge+%28j+%5Cneq+j%27%29+%5Cwedge+%5Cleft%28+%5B%7C+i%3B+j+%7C%5D+%5Ccap+%5B%7C+i%27%2C+j%27+%7C%5D+%3D+%5Cleft%5C%7B+%5Cbegin%7Barray%7D%7Bl%7D+%5Cemptyset+%5C%5C+%5B%7C+i%3B+j+%7C%5D+%5C%5C+%5B%7C+i%27%3B+j%27+%7C%5D+%5Cend%7Barray%7D+%5Cright.+%5Cright%29+&bg=ffffff&fg=000&s=0&c=20201002)

C'est bien beau comme problème, mais, en vrai comment on trouve une solution ? L'idée générale c'est que toute super structure peut se décomposer sous la forme explicitée Figure 8 :

Comment le problème est résolu ? Je vais pas vous donner l'algorithme (ça sera le sujet d'un autre article ^^) mais c'est un problème soluble et pas si difficilement que ça. Et puis, je vous donnerai du code qui fait ce qu'il faut.

Bien sur, sur cet exemple artificiel (Figure 9) la planarisation a l'air d'enlever tous les arcs, ce n'est pas le cas.
Ça sort juste la plus grande structure planaire possible. et c'est ce dont on a besoin pour représenter la structure sous forme dot-parenthesis ou d'arbre pour faire de l'alignement.
Quelles informations intéressantes pour la structure des ARN peut-on extraire d'un fichier PDB ?
À partir des coordonnées atomiques contenues dans un fichier PDB, il est possible d'extraire plusieurs types d'informations. Nous allons nous intéresser à 2 types d'informations :
- les angles dièdres
- les interactions entre bases
Si les interactions entre bases sont nécessaires pour trouver les structures secondaires des ARN, les angles dièdres pas du tout. Je vous présente les angles dièdres parce que c'est un sujet que je trouve intéressant, qui peut être utile quand on travaille sur la structure 3D des ARN et parce que le code que j'ai écrit et que je présente à la fin permet de les extraire facilement, du coup, qui peut le plus, peut le moins.
Et si on parlait d'angles dièdres ?
Ça n'est probablement pas une surprise pour vous, mais dans la PDB il n'y a pas que des structures de protéines mais aussi des structures d'acides nucléiques et, en particulier, tout un tas de structures d'ARN, que nous allons utiliser par la suite.
Dans ces fichiers qui, pour rappel, contiennent les coordonnées atomique d'une molécule (en l'occurence, des chaînes d'ARN), peuvent servir à extraire tout un tas de données et on va se concentrer d'abord sur les angles dièdres
Mais qu'est ce qu'un angle dièdre ?
Définition 6 : Angle dièdre

En chimie, un angle dièdre est l'angle formé entre 2 plans définis par 3 atomes, ces 2 plans ayant 2 atomes en commun.
Les angles dièdres de l'ARN
Maintenant que vous vous rappelez ce qu'est un angle dièdre, passons aux angles dièdres dans l'ARN, et il y en a un paquet ! La Figure 11 montre les angles dièdres de l'ARN.

Comme vous l'avez sûrement constaté, sur la Figure 11, la définition de l'angle χ est un peu floue. C'est normal : cet angle dépend de la base, de selon si elle est purine ou pyrimidine.
Pour plus de clareté, je vous livre Figure 12 la liste des atomes mis en jeu pour calculer les angles dièdres.
|
1 2 3 4 5 6 7 8 9 10 |
alpha (α): O3'(i-1) - P(i) - O5'(i) - C5'(i) beta (β): P(i) - O5'(i) - C5'(i) - C4'(i) gamma (γ): O5'(i) - C5'(i) - C4'(i) - C3'(i) delta (δ): C5'(i) - C4'(i) - C3'(i) - O3'(i) epsilon (ε): C4'(i) - C3'(i) - O3'(i) - P(i+1) zeta (ζ): C3'(i) - O3'(i) - P(i+1) - O5'(i+1) chi (χ): O4'(i) - C1'(i) - N1(i) - C2(i) -- pour les pyrimidines chi (χ): O4'(i) - C1'(i) - N9(i) - C4(i) -- pour les purines eta (η): C4'(i-1) - P(i) - C4'(i) - P(i+1) teta (θ): P(i) - C4'(i) - P(i+1) - C4'(i+1) |
Figure 12 : définition exacte des angles impliqués dans les différents angles dièdres et de torsions dans l'ARN.
Vous avez peut être remarqué qu'il y a 2 angles supplémentaires Figure 12 dont je n'ai pas encore parlé, η et θ.
Ces 2 angles ont été intégré par Olson [3] afin d'avoir une mesure de la flexibilité inter-base des ARN (et ça marche !).
Et les types d'interactions ?
En vrai, ce qui nous intéresse pour la structure secondaire des ARN, c'est les interactions et les types d'interactions plus que les angles dièdres (ce n'était qu'une partie bonus que j'aime bien).
Quand on parle de type d'interactions, quand on a fait un peu de structure dans sa vie, logiquement on pense aux :
- interactions d'empilement (i.e. de stacking)
- interactions bord-à-bord (i.e. edge-to-edge)
Là, je vais me focaliser sur les interactions bord-à-bord existant au travers de liaison hydrogène (et ça en fait déjà un paquet !).
Je suppose que vous avez tous entendu parler des intéractions canoniques de type Watson-Crick/Watson-Crick mais, en réalité, elles sont loin d'être les seules intéractions existantes. En effet, dans la classification de Léontis et Westhof, il existe 12 types d'intéractions possible, comme on peut le voir Figure 14.

Petit retour en arrière : d'où viennent ces 12 types ? Qu'est ce que c'est que cette arnaque ?
C'est pas une arnaque du tout, c'est juste que, contrairement à ce que vous pouviez croire, les interactions entre bases de l'ARN ne sont pas exclusivement entre les « bords » de Watson-Crick (cf Figure 13) mais aussi entre les « bords » de Hoogsten et les « bords » Sugar. Ce qui nous fait 3 « bords » d'intéractions qui peuvent tous (on va dire ça pour l'instant) intéragir les uns avec les autres !
On peut très bien imaginer une adénine dont le bord Watson-Crick intéragit avec le bord Hoogsten d'une uracile par exemple.
C'est le moment où vous me dites que 
C'est parce qu'il reste l'inclinaison du lien qui peut être cis ou trans, comme cela est expliqué sur un schéma Figure 13, ce qui nous amène bien à 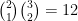
Celles ci sont décrites, pour plus de clareté sur la Figure 14.

En pratique, 11 sur les 12 familles d'intéractions possibles ont été déterminées dans des structures de la PDB, la famille manquante étant la famille cis Hoogsten/Hoogsten (à vous de jouer =p).
C'est bien beau tout ça, mais comment on récupère tout ça ?
Il est possible d'obtenir les structures arcs-annotées d'un ARN à partir d'un fichier PDB grâce au logiciel rnaview.
En guise de conclusion, je vais vous donner un peu de code qui vous permettra de jouer avec les notions que l'on a vue dans l'article.
Cela vous permettra d'extraire des structures bien parenthésées (donc des structures secondaires) et de regarder les interactions préservées et celles rejetées par la planarisation (et pourquoi pas d'en faire des stats, j'en ai fait pendant mon stage, c'est surprenant) mais aussi de récupérer les angles dièdres et de regarder leurs distributions (je l'ai fait aussi, n'oubliez pas votre trigonométrie de base).
Je comprends complètement que vous ayez autre chose à faire de vos journées, mais si jamais vous en avez besoin, vous savez qu'il y a un code tout fait qui vous attend. (Et si vous trouvez des bugs \o/ dites-le dans les commentaires, je patcherai)
Le code python disponible ici, sa documentation est là et vous pouvez télécharger le paquet pip à cet endroit et juste taper
pip install pprna-1.0.0b1.tar.gz.
Des exemples de ce code sont disponibles ici :
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 |
import csv import re import sys import os sys.path.append("../python/RNA") import RNAParser import RNASecondaryStructure fieldnames = ["PDB.ID", "Full.ID", "Length", "Total", "Structure method", "Resolution", "K_minus_cis", "K_plus_cis","K_W_W_cis", "K_W_W_tran", "K_H_H_cis", "K_H_H_tran", "K_S_S_cis", "K_S_S_tran", "K_W_S_cis", "K_W_S_tran", "K_W_H_cis", "K_W_H_tran", "K_H_S_cis", "K_H_S_tran", "D_minus_cis", "D_plus_cis", "D_W_W_cis", "D_W_W_tran", "D_H_H_cis", "D_H_H_tran", "D_S_S_cis", "D_S_S_tran", "D_W_S_cis", "D_W_S_tran", "D_W_H_cis", "D_W_H_tran", "D_H_S_cis", "D_H_S_tran"] keys = [ [("-/-", "cis")], [("+/+", "cis")], [("W/W", "cis")], [("W/W", "tran")], [("H/H", "cis")], [("H/H", "tran")], [("S/S", "cis")], [("S/S", "tran")], [("W/S", "cis"), ("S/W", "cis")], [("W/S", "tran"), ("S/W", "tran")], [("W/H", "cis"), ("H/W", "cis")], [("W/H", "tran"), ("H/W", "tran")], [("H/S", "cis"), ("S/H", "cis")], [("H/S", "tran"), ("S/H", "tran")], ] with open("/home/chopopope/PDB-RNA/interactions_data.csv", "wb") as csvfile : # Instanciate writer and writes headers. writer = csv.DictWriter(csvfile, fieldnames=fieldnames) writer.writeheader() for file in os.listdir("~/PDB-RNA/"): # For every pdb file # Re-initate RNA PDB parser parser = RNAParser.PDBParser(annotation=True) pdb_filename = os.path.join("~/PDB-RNA/", file) # Retrieve the PDB ID (i.e. the file name) PDB_ID = re.split("/", pdb_filename)[-1].strip(".pdb") # Initiate structure structure = parser.get_structure(file, pdb_filename) # Retrieve data from header METHOD = structure.header["structure_method"] RESOLUTION = structure.header["resolution"] for model in structure : # Each structure may have several models for chain in model.child_list : # Each model may have several chains # Retrieves the full-id FULL_ID = PDB_ID + "_" + str(model.id) + "_" + chain.id # Retrieves sequence length (useful for stats) LENGTH = len(chain.get_sequence()) # Computes the secondary structure plop = RNASecondaryStructure.RNASecondaryStructure(chain) # Initialization of the dict that will hold the data to store # in a # csv file dico = {} # Let's start with the easy fields dico[fieldnames[0]] = PDB_ID dico[fieldnames[1]] = FULL_ID dico[fieldnames[2]] = LENGTH dico[fieldnames[4]] = METHOD dico[fieldnames[5]] = RESOLUTION # Now, let's get to work ! for key, field in zip(keys, fieldnames[6 :20]): # i.e. all the "kept" fields dico[field] = plop.kept_interactions().get(key[0], 0) if len(key) > [1]: # i.e. double interaction dico[field] += plop.kept_interactions().get(key[1], 0) # Now, let's get to work ! for key, field in zip(keys, fieldnames[20 :]): # i.e. all the "discarded" fields dico[field] = plop.discarded_interactions().get(key[0], 0) if len(key) > [1]: # i.e. double interaction dico[field] += plop.discarded_interactions().get(key[1], 0) dico[fieldnames[3]] = sum([dico[i] for i in fieldnames[6 :]]) # Writes the dict to the csv file. writer.writerow(dico) csvfile.close() |
Exemple de code 1 : Récupération des intéractions conservées et supprimées de chaque modèle de chaque chaîne de chaque structure d'un dossier contenant des PDB d'ARN dans un fichier CSV.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 |
#!/usr/bin/env python2.7 # -*- coding:utf-8 -*- # This code loads every provided .pdb files (with RNA) and process each of them # so that we can retrieve the dihedral angles of every single nucleotide # chain of each model of each structure and store them in a csv file. # # PDB_ID | Full_ID | Length | Res_ID | Structure_method | Resolution | alpha | […] import csv import re import sys import os sys.path.append("../python/RNA") import RNAParser import time fieldnames = ["PDB ID", "Full ID", "Length", "Residue ID", "Structure method", "Resolution", "alpha", "beta", "chi", "delta", "epsilon", "eta", "gamma","theta", "zeta"] with open("~/PDB-RNA/dihedrals_data.csv", "wb") as csvfile : # Instanciate writer and writes headers. writer = csv.DictWriter(csvfile, fieldnames=fieldnames, quoting=csv.QUOTE_NONNUMERIC) writer.writeheader() for file in os.listdir("~/PDB-RNA/"): # For every pdb file # Re-initate RNA PDB parser parser = RNAParser.PDBParser(annotation=True) pdb_filename = os.path.join("~/PDB-RNA/", file) # Retrieve the PDB ID (i.e. the file name) PDB_ID = re.split("/", pdb_filename)[-1].replace(".pdb", "") # Initiate structure structure = parser.get_structure(file, pdb_filename) # Retrieve data from header METHOD = structure.header["structure_method"] RESOLUTION = structure.header["resolution"] for model in structure : # Each structure may have several models for chain in model.child_list : written = False # Each model may have several chains # Retrieves the full-id FULL_ID = PDB_ID + "_" + str(model.id) + "_" + chain.id # Retrieves sequence length (useful for stats) LENGTH = len(chain.get_sequence()) data = chain.get_dihedrals() for residue in data.keys(): # Retrieve the esidue ID RES_ID = residue.get_id()[1] # Initialization of the dict that will hold the data to store in a # csv file dico = {} # Let's start with the easy fields dico[fieldnames[0]] = PDB_ID dico[fieldnames[1]] = FULL_ID dico[fieldnames[2]] = LENGTH dico[fieldnames[3]] = RES_ID dico[fieldnames[4]] = METHOD dico[fieldnames[5]] = RESOLUTION for field in fieldnames[6 :]: # Let's work on the angles dico[field] = data[residue][field] # Writes the dict to the csv file. writer.writerow(dico) time.sleep(0.1) csvfile.close() |
Exemple de code 2 : Récupération des angles dièdres de chaque modèle de chaque chaîne de chaque structure d'un dossier contenant des PDB d'ARN dans un fichier CSV.
Merci à ZaZo0o, Kumquatum, Clem_ et lelouar pour les commentaires, corrections, la détection de fautes de français et les coups de pieds au cul !
[1]: Introduction to RNA secondary structure comparison, Schirmer, Stefanie and Ponty, Yann and Giegerich, Robert, Humana Press, 2014
[2]: VARNA : Interactive drawing and editing of the RNA secondary structure, Darty, Kévin and Denise, Alain and Ponty, Yann, Bioinformatics, 2009
[3]: Configurational statistics of polynucleotide chains. A single virtual bond treatment., Olson, W. K., Macromolecules, 1975
[4]: Geometric nomenclature and classification of RNA base pairs, Leontis, Neocles B. and Westhof, Eric, RNA, 2001
[5]: The PyMOL Molecular Graphics System, Schrödinger, LLC, 2010
Laisser un commentaire