

Étiquette : script
-
Jouer avec l'API de KEGG

Il n'est pas rare que nous ayons un jour besoin de récupérer des informations de la base de données KEGG (Kyoto Encyclopedia of Genes and Genomes). Cette base de données fournit un nombre conséquent d'informations sur les génomes et les réseaux de gènes mais également sur les voies métaboliques ou les maladies. Dans ces cas…
-
Ajoutez une interface graphique à votre script en 4 lignes avec Gooey
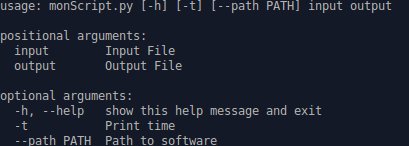
Vous venez de terminer votre analyse bio-informatique. Pour cette dernière, vous avez réalisé un script qui pour l'instant, il faut le dire, n'est pas du tout réutilisable par une tierce personne. Même vous dans 6 mois vous n'êtes pas sûr de vous souvenir de ce que vous avez fait. Pourtant, l'un des intérêts de la…
-
Parser des fichiers HTML en Python

Langage : PythonBibliothèques : bioservices, HTMLParser, re (partiellement)Niveau : débutant-intermédiaire Dans un article précédent, je vous ai présenté le module bioservices en Python. Au cours de mon travail j'ai été amenée à récupérer des informations sur les termes Gene Ontology, et notamment sur les relations entre différents termes. Cependant, les formats de fichiers récupérés sont différents en fonction…
-
Soirée BED & FASTA !

Après la petite histoire de l’analyse des séquences d’ADN, voici un tutoriel pour apprendre quelques trucs et astuces dans ce domaine. Biologiste en mal de connaissances de programmation ou pro de R, vous trouverez ici de quoi vous amuser avec un fichier Fasta ou un Bed. Nous allons voir comment faire un alignement multiple de…
-
Bioservices, un module Python très utile

Dans notre domaine si vaste, il existe de nombreuses bases de données (cf. Bases de données, notions par nahoy), et parmi ces bases, un certain nombre d'entre elles propose un service web pour accéder à leurs données à partir d'un script. Le problème principal qui peut nous freiner, ou nous faire peur, lorsque l'on se…
-
Astuce : ajouter des options dans un script Bash avec getopt
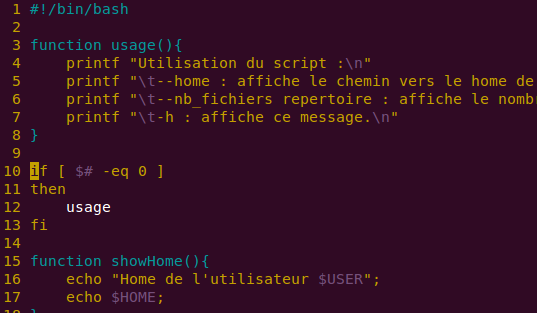
But : comprendre le fonctionnement de getopt en Bash pour éviter la multiplications de script là où un seul générique pourrait suffire. Prérequis : savoir faire des scripts Bash, connaître la substitution de commande et savoir manipuler les arguments. Difficulté : 2 (moyen) Pour ceux qui codent en Perl, vous connaissez déjà sûrement le module GetOpt et plus…
-
À la découverte de BioMart !

Dans le cadre de mon travail, j'ai récemment découvert un outil formidable pour la consultation et la récupération de données à partir de certaines banques : BioMart. Après avoir passé la frustration de devoir utiliser l'interface du service fourni pour télécharger les différentes données dont j'avais besoin, je me suis renseignée davantage sur ce logiciel. De…
-
Récupérer la fiche d'un gène avec les Eutils du NCBI
En bioinformatique il n'est pas rare que l'on ait besoin d’accéder à des informations disponibles sur des bases de données internationales, nous verrons ici le cas de la banque Gene du NCBI. Avant de s'intéresser à la récupération d'une fiche d'un gène en passant par les Eutils, un peu de théorie et d'explications sur une fiche…