

Introduction
Dans le domaine de la bioinformatique l'alignement de séquences est une opération courante pour comparer des séquences biologiques (ADN, ARN, protéines). Nous avons vu dans un précédent article le principe de l'algorithme de Needleman-Wunsch qui produit un alignement global optimal de deux séquences.
Au-delà de l'alignement par paire, l'alignement multiple de séquences (Multiple Sequence Alignment ou MSA) permet de comparer simultanément plusieurs séquences afin de mettre en évidence des régions conservées, de repérer des motifs fonctionnels ou de faciliter des analyses phylogénétiques. La généralisation naïve de l'algorithme de Needleman-Wunsch pour plus de deux séquences est malheureusement irréalisable à cause de sa complexité temporelle en 
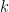
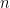
Dans ce billet, je vais vous présenter l'intérêt et le principe de l'alignement multiple en étoile (Star Alignment ou parfois Center Star Alignment), une approche qui repose sur la réalisation d'alignements par paires, puis leur fusion autour d'une séquence centrale de référence.
1. Exemple d'incohérences entre alignements de paires
L'une des raisons qui rend le MSA si complexe est le fait que les alignements de paires de séquences, bien qu'optimaux lorsqu'on les considère individuellement, sont souvent incohérents entre eux. Il n'est donc pas possible de fusionner simplement ces alignements pour produire un alignement multiple.
Prenons un exemple simple de 3 séquences de 10 nucléotides pour montrer comment se manifestent ces incohérences. Par commodité je choisis les paramètres d'alignement suivants : +1 pour une identité (match), -1 pour une substitution (mismatch) et -1 également pour une lacune (gap). Avec les 3 séquences (seq1, seq2, seq3) suivantes nous obtenons 3 paires d'alignements (ali1/2, ali1/3, ali2/3).
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 |
seq1 = AATAGGGCTC seq2 = GAATAGTTGG seq3 = GTGGCTGTCT ali1/2 = 01234 56789 -AATAG--GGCTC GAATAGTTGG--- 0123456789 ali1/3 = 01234567 89 AATAGGGC--TC- -GT--GGCTGTCT 01 23456789 ali2/3 = 012345 6 789 GAATAG-T-TGG G--TGGCTGTCT 0 123456789 |
Pour faciliter la comparaison entre ces 3 alignements et mettre en évidence leurs incohérences on peut numéroter les nucléotides. Ainsi :
- D'après l'alignement 1, le nucléotide 1 de la séquence 1 (le deuxième A) est homologue au nucléotide 2 de la séquence 2.
- D'après l'alignement 2, ce même nucléotide 1 de la séquence 1 est homologue au nucléotide 0 de la séquence 3.
On devrait donc en déduire que le nucléotide 2 de la séquence 2 et le nucléotide 0 de la séquence 3 sont homologues. L'homologie est en effet une relation transitive. Mais nous constatons au contraire que dans l'alignement 3 le nucléotide 2 de la séquence 2 n'est homologue à rien du tout dans la séquence 3, et que le nucléotide 0 de la séquence 3 est homologue au nucléotide 0 de la séquence 2.
Les 3 alignements pris ensemble sont donc incohérents, on ne peut pas les fusionner en un alignement multiple unique sans contredire certaines portions de ces alignements par paires.
2. Principe de l'alignement en étoile
Dans l'alignement en étoile on commence par choisir l'une des séquences qu'on désignera comme centrale et qui servira de référence. On aligne cette séquence successivement avec toutes les autres, mais on n'aligne pas ces autres séquences entre elles.
Schématiquement, on peut donc placer la séquence centrale au milieu d'une étoile dont toutes les branches représentent un alignement par paire avec l'une des autres séquences (voir schéma ci-dessous). Le fait de ne pas considérer les alignements entre les séquences de la périphérie de l'étoile permet d'éliminer les incohérences, mais on perd aussi bien sûr de l'information.

3. Choisir une séquence centrale
Il existe plusieurs stratégies pour choisir la séquence centrale de manière à minimiser cette perte d'information. La plus commune est de choisir la séquence qui est en moyenne la plus proche de toutes les autres. On espère alors de cette manière privilégier les alignements par paire contenant le moins d'erreurs d'alignement.
Plusieurs détails importants peuvent varier dans la mise en œuvre de cette stratégie. Par exemple quelle formule on décide d'appliquer pour calculer les distances entre les séquences. Pénalise-t-on ou ignore-t-on les gaps ? Est-ce qu'on utilise une distance brute comme la p-distance ou est-ce qu'on applique une correction comme avec la distance de Jukes-Cantor ? Comment calcule-t-on cette moyenne des distances ? Avec ou sans pondération ? Ou peut-être préfèrera-t-on utiliser la médiane finalement ?
Bref, restons simple et poursuivons avec notre exemple. L'alignement 1 montre que les séquences 1 et 2 ont une p-distance nulle car il n'y a que des matchs et aucun mismatchs. Le calcul de la p-distance consiste en effet à diviser le nombre de nucléotides différents par le nombre total de nucléotides comparés, ce qui implique d'ignorer toutes les positions avec des gaps (le "p" signifie "proportion").
L'alignement 2 montre un seul mismatch sur 7 positions sans gap, donc la p-distance est de 1/7 entre les séquences 1 et 3. Enfin l'alignement 3 montre 3 mismatchs sur 8 positions sans gap, la distance entre les séquences 2 et 3 est donc de 3/8. Cela nous permet de calculer la distance moyenne d'une séquence aux deux autres séquences :
|
1 2 3 4 5 6 |
dist(seq1, seq2) = 0 dist(seq1, seq3) = 1/7 dist(seq2, seq3) = 3/8 mean_dist(seq1) = (0 + 1/7) / 2 = 1/14 = 0.0714 mean_dist(seq2) = (0 + 3/8) / 2 = 3/16 = 0.1875 mean_dist(seq3) = (1/7 + 3/8) / 2 = 29/112 = 0.2589 |
La séquence 1 est celle qui apparait en moyenne la plus proche, c'est donc elle qu'on choisit ici comme séquence centrale. On rejette donc complètement l'alignement 3 qui n'inclut pas la séquence centrale.
4. Construire une séquence centrale cohérente
Dans une implémentation simple du principe de l'alignement en étoile, l'étape qui suit le choix de la séquence centrale est celui de l'agglomération de toutes les versions de cette séquence pour construire une unique version de référence. Cette version finale doit incorporer toutes les lacunes (gaps) des alignements par paires retenus. Voici les deux versions de la séquence centrale de notre exemple provenant respectivement des alignements 1 et 2 :
|
1 2 |
seq1v1 = -AATAG--GGCTC seq1v2 = AATAGGGC--TC- |
Pour agglomérer deux versions il suffit de les parcourir simultanément avec deux pointeurs différents. Ici je parle de pointeur par abus de langage pour désigner simplement une variable qui contient la position qu'on lit dans une chaine de caractère (on peut accélérer l'algorithme si on utilise un vrai pointeur qui contient l'adresse mémoire de cette position mais je ne rentrerai pas dans ces détails d'optimisation). On procède alors comme suit :
- Lorsqu'on lit avec les deux pointeurs un même caractère non-gap alors on ajoute ce caractère à la nouvelle version et on incrémente les deux pointeurs. Remarquez que ce caractère non-gap est nécessairement le même dans les deux séquences, si ce n'est pas le cas c'est qu'il y a une erreur dans l'implémentation de l'algorithme.
- Lorsqu'on lit un gap avec les deux pointeurs alors on incrémente de la même façon les deux pointeurs et on ajoute un gap à la nouvelle version.
- Lorsqu'on lit avec un seul des deux pointeurs un gap alors on ajoute ce gap à la nouvelle version et on incrémente seulement ce pointeur, mais pas l'autre.
Voici ce que cela donne sur notre exemple :
- Le pointeur p1 et le pointeur p2 sont initialisés à 0 et lisent respectivement "-" et "A". Seul le pointeur p1 est donc incrémenté et on ajoute un gap à la nouvelle version.

- Le pointeur p1 vaut 1 et p2 vaut toujours 0, les deux lisent "A", donc "A" est ajouté à la nouvelle version et les deux sont incrémentés.

- De la même manière on ajoute "A", "T", "A", "G". On a alors p1 = 6 et p2 = 5, p1 lit "-" et p2 lit "G", on ajoute alors un gap à la nouvelle version et seul p1 est incrémenté à 7.

- Avec p1 = 7 et p2 = 5 on lit respectivement encore "-" et "G", donc on ajoute à nouveau un gap et on incrémente seulement p1.
- Avec p1 = 8 et p2 = 5 on lit deux "G", donc on l'ajoute et on incrémente les deux. Avec p1 = 9 et p2 = 6 on lit encore deux "G" donc on fait de même. Ensuite avec p1 = 10 et p2 = 7 on lit deux "C", donc on l'ajoute et on incrémente les deux.

- Avec p1 = 11 et p2 = 8 on lit respectivement "T" et "-", donc cette fois c'est seulement p2 qu'on incrémente à 9 et on ajoute un gap.

- Je vous laisse finir ?
Voici le résultat qu'on obtient, vous pouvez constater que cet algorithme permet bien d'incorporer les gaps des deux versions dans une seule nouvelle version de la séquence :
|
1 |
seq1vf = -AATAG--GGC--TC- |
Si on a plus de deux versions à fusionner, un algorithme naïf consiste à tout simplement fusionner ces versions progressivement dans une boucle. L'ordre des fusions n'a aucune influence sur le résultat final.
5. Mettre en cohérence toutes les autres séquences
À cette étape nous connaissons la version finale de la séquence centrale et nous avons aussi différentes versions locales de cette séquence centrale alignées avec chacune des autres séquences. Ce sont ces autres séquences que nous voulons maintenant aligner avec la version finale de la séquence centrale.
Le principe est le même que dans l'étape précédente. Les gaps manquants dans ces autres séquences sont les mêmes que ceux qui manquent si on compare la version finale de la séquence centrale avec sa version locale pour une paire donnée. Il suffit donc de parcourir chaque alignement et de comparer les deux versions de la séquence centrale : la finale et la locale.
- Lorsque les deux versions de la séquence centrale ont le même caractère (gap ou non) alors on ne modifie pas la séquence à aligner, puis on incrémente les deux pointeurs de lecture.
- Lorsque les deux versions sont différentes alors on ajoute un gap à cette position dans la séquence à aligner, puis on incrémente seulement le pointeur de lecture de la version finale de la séquence centrale.
Avec cette procédure on obtient l'alignement multiple final suivant :
|
1 2 3 |
seq1vfa : -AATAG--GGC--TC- seq2vfa : GAATAGTTGG------ seq3vfa : --GT----GGCTGTCT |
Comme attendu, cet alignement multiple est parfaitement cohérent avec les deux alignements par paire ali1/2 et ali1/3, mais n'est que partiellement cohérent avec l'alignement ali2/3. En fait si on compare ces alignements plus précisément, on ne retrouve en commun dans l'alignement multiple final et dans l'alignement ali2/3 que le premier "T" (nucléotide d'indice 3 de la séquence 2 et nucléotide d'indice 1 de la séquence 3), aucunes des autres associations ne sont conservées.
6. Évaluation de la qualité de l'alignement multiple
Comme on l'a vu, le fait d'avoir choisi la séquence 1 comme séquence centrale nous a obligé à ignorer les informations de l'alignement ali2/3. Si on avait choisi la séquence 2 ou bien la séquence 3 comme séquence centrale alors nous aurions ignoré à la place d'autres informations et nous aurions obtenu les alignements multiples suivants.
Alignement multiple en étoile avec la séquence 2 au centre :
|
1 2 3 |
seq1vfb : -AATAG----GGCTC seq2vfb : GAATAG-T-TGG--- seq3vfb : G--TGGCTGTCT--- |
Alignement multiple en étoile avec la séquence 3 au centre :
|
1 2 3 |
seq1vfc : AA--TAGG--G-CTC seq2vfc : -GAAT-AG-T-TGG- seq3vfc : -G--T-GGCTGTCT- |
Comment savoir si un de ces alignements ne serait pas meilleur que celui que nous avons obtenu ?
Il existe plusieurs manières d'évaluer la qualité d'un alignement multiple. On fait généralement appel au calcul d'une fonction objective qu'on cherche à minimiser ou à maximiser selon les cas. Je rappelle que le but est d'obtenir un maximum de vraies positions homologues et un minimum d'erreurs. Il est donc naturel d'extrapoler l'utilisation du score d'alignement qui a fait le succès de l'algorithme de Needleman-Wunsch pour inférer correctement les positions homologues d'une paire de séquences.
On peut donc calculer un score d'alignement multiple en faisant la somme des scores d'alignement pour chaque paire de cet alignement multiple. Cette fonction objective s'appelle sum-of-pairs ou "somme des paires". On doit bien sûr ignorer lors de ce calcul les paires de positions avec deux gaps.
Commençons par calculer le score pour un alignement simple :
|
1 2 3 |
seq1vfa : -AATAG--GGC--TC- seq2vfa : GAATAGTTGG------ eval : .+++++..++.00..0 |
Sur la ligne d'évaluation j'ai mis des points . pour représenter les mismatchs et les gaps (par simplicité tous les deux ont ici un score de -1), les plus + représentent les matchs (ici avec un score de +1), et j'ai mis des zéros 0 pour deux gaps. On compte 7 plus et 6 points, donc le score de cet alignement est de 7 - 6 = 1.
On fait la même chose pour les deux autres paires de l'alignement multiple A (celui avec la séquence 1 centrale) on obtient de cette manière le score de -10 :
|
1 2 3 4 |
score(seq1vfa, seq2vfa) = 1 score(seq1vfa, seq3vfa) = -1 score(seq2vfa, seq3vfa) = -10 scorea = -10 |
Sans surprise c'est la paire 2/3 qui reçoit le score le plus bas puisque ses informations ont été ignorées. Les alignements B et C reçoivent respectivement les scores -12 et -13 :
|
1 2 3 4 |
score(seq1vfb, seq2vfb) = 1 score(seq1vfb, seq3vfb) = -11 score(seq2vfb, seq3vfb) = -2 scoreb = -12 |
|
1 2 3 4 |
score(seq1vfc, seq2vfc) = -10 score(seq1vfc, seq3vfc) = -1 score(seq2vfc, seq3vfc) = -2 scorec = -13 |
Ces scores plus bas nous montrent que ces alignements alternatifs ne sont pas meilleurs et que nous avons eu raison de choisir la séquence 1 comme séquence centrale.
Notez bien que des choix de paramètres différents pour les matchs, les mismatchs et les gaps changeraient les alignements par paires et l'alignement multiple final en modifiant les calculs des scores. On peut de plus choisir une autre fonction objective que la simple sum-of-pairs, à commencer par la weighted sum-of-pairs qui ajoute une pondération plus ou moins complexe dans le calcul de cette somme.
7. Intérêts et limites
L'alignement multiple en étoile est simple à implémenter et très rapide à mettre en œuvre. En fait ce sont les alignements par paires qui constituent le goulot d'étranglement principal de l'algorithme. Mais on peut éviter d'avoir à calculer les alignements de toutes les paires si notre stratégie de choix de la séquence centrale ne le requiert pas (je ne détaillerai pas ici les techniques de calculs de distances entre séquences basées sur les k-mères qui ne nécessitent pas d'alignements).
Malheureusement il n'existe pas de stratégie de choix de la séquence centrale qui nous assure d'obtenir le meilleur alignement multiple. On pourrait être tenté d'essayer toutes les séquences centrales possibles et de choisir l'alignement final ayant le meilleur score, mais cette méthode est beaucoup plus lente car elle nous oblige à calculer tous les alignements par paires.
Même avec une stratégie aussi couteuse l'alignement en étoile est peu performant avec des séquences assez éloignées. Les résultats ne sont fiables que pour des séquences très proches. Cela est dû au fait que l'algorithme ignore une grande quantité d'informations dans les alignements par paires qui ont été rejetés. Ainsi, une erreur d'alignement commise dans l'un des alignements par paire retenu ne peut pas être corrigée par les informations contenues dans les alignements par paires rejetés.
Des stratégies d'alignement multiple plus complexes et plus précises ont donc été développées pour exploiter au maximum l'ensemble des informations disponibles. Les alignements multiples basés sur la cohérence (consistency based MSA) essaient par exemple de la cohérence des homologies entre tous les alignements par paires. Les alignements multiples progressifs (progressive MSA) essaient plutôt de construire un alignement en ajoutant les séquences une par une et en l'alignant de manière à prendre en compte les informations dans toutes les séquences déjà alignées.
Références
- Gusfield, D. (1997). Multiple string comparison - the Holy Grail. Algorithms on Strings, Trees and Sequences, 332-366.
- Gusfield, D. (1993). Efficient methods for multiple sequence alignment with guaranteed error bounds. Bulletin of mathematical biology, 55(1), 141-154.
- Gusfield, D. (1991). Efficient algorithms for inferring evolutionary trees. Networks, 21(1), 19-28.
Annexe : implémentation de l'alignement en étoile en Python
L'algorithme d'alignement en étoile suivant n'est pas du tout optimisé. Cette manière d'écrire le code permet de séparer clairement les étapes pour que l'ensemble soit facilement compréhensible, mais cela provoque des redondances. Référez-vous à l'article précédent pour l'implémentation de l'algorithme de Needlman-Wunsch qui produit les alignements par paires.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 |
def star_alignment(sequences, center) : # Liste d'alignements par paires avec la séquence centrale en première position pairs = [] for i in range(len(sequences)) : if i != center : _, new_center, new_seq = needleman_wunsch(sequences[center], sequences[i], 1, -1, -1) pairs.append([new_center, new_seq]) # Mise en cohérence des différentes versions de la séquence centrale curr_center = pairs[0][0] for i in range(1, len(pairs)) : p1 = 0 # Pointeur pour lire la version courante de la séquence centrale p2 = 0 # Pointeur pour lire la version suivante dans la liste next_center = pairs[i][0] new_center = "" # Construction d'une nouvelle version de la séquence centrale while p1 < len(curr_center) or p2 < len(next_center) : if p1 == len(curr_center) : c1 = "" # Attention aux limites de dépassement des chaines de caractères else : c1 = curr_center[p1] if p2 == len(next_center) : c2 = "" # Idem else : c2 = next_center[p2] if c1 == c2 : new_center += c1 p1 += 1 p2 += 1 elif c1 == "-" : p1 += 1 new_center += "-" else : p2 += 1 new_center += "-" curr_center = new_center # Mise à jour de la version courante de la séquence centrale # Mise en cohérence de chaque séquence avec la version finale de la séquence centrale alignment = [] for i in range(len(pairs)) : p1 = 0 # Pointeur pour lire les caractères de la version finale de la séquence centrale p2 = 0 # Pointeur pour lire les caractères de la version locale de la séquence dans chaque paire next_center = pairs[i][0] # Version locale de la séquence centrale next_seq = pairs[i][1] # Séquence à aligner new_seq = "" # Séquence alignée while p1 < len(curr_center) or p2 < len(next_center) : if p1 == len(curr_center) : c1 = "" # Attention aux limites de dépassement des chaines de caractères else : c1 = curr_center[p1] if p2 == len(next_center) : c2 = "" # Idem else : c2 = next_center[p2] if c1 == c2 : new_seq += next_seq[p2] p1 += 1 p2 += 1 else : new_seq += "-" p1 += 1 alignment.append(new_seq) # Placement de la séquence centrale au bon endroit dans la liste des séquences alignées alignment.insert(center, curr_center) return alignment def sumofpairs(sequences) : score = 0 for i in range(len(sequences) - 1) : for j in range(i + 1, len(sequences)) : seqA = sequences[i] seqB = sequences[j] pair = 0 for k in range(len(seqA)) : if seqA[k] == seqB[k] and seqA[k] != "-" : pair += 1 elif seqA[k] != seqB[k] : pair -= 1 score += pair return score # Exemple d'utilisation simple seq1 = "AATAGGGCTC" seq2 = "GAATAGTTGG" seq3 = "GTGGCTGTCT" sequences = [seq1, seq2, seq3] alignment = star_alignment(sequences, 0) score = sumofpairs(alignment) for i in range(len(sequences)): print("Séquence " + str(i + 1) + " :", sequences[i]) print("") for i in range(len(alignment)): print("Séquence " + str(i + 1) + " :", alignment[i]) print("Score d'alignement multiple :", score) |
Merci à Léopold Carron et Lee Mariault pour la relecture.
Laisser un commentaire