


Introduction
L'analyse des séquences biologiques (ADN, ARN ou protéines) est un sujet central de la biologie moléculaire et de la bioinformatique. Dans ce cadre, chercher à savoir à quel point deux séquences sont similaires ou dissimilaires est une des questions qu'on se pose le plus souvent (par exemple dans le but d'analyser des relations phylogénétiques). Alors comment fait-on pour quantifier cette dissimilarité ?
La dissimilarité entre deux séquences peut se mesurer simplement par le nombre minimal de transformations qui sont nécessaires pour passer de l'une à l'autre. On pourrait donc compter les différences position par position. Disons que l'on ait les deux séquences de 11 nucléotides suivantes :
|
1 2 |
AGCGTACATGC ACGCACGTCAC |
Combien de différences comptez-vous ? Position par position on compte 9 différences. En fait, seuls le premier et le dernier nucléotides sont identiques. Le problème est que si nous décalons les séquences, comme ci-dessous, nous obtenons un résultat différent.
|
1 2 |
AGCGTACATGC- -ACGCACGTCAC |
Même en comptant les deux lacunes (ou gaps) représentées par des tirets comme des différences, cet alignement n'en comporte plus que 7. Ici nous n'avons fait que décaler la totalité de la deuxième séquence d'un cran par rapport à la première, mais nous pourrions aussi placer des lacunes à l'intérieur des séquences pour améliorer l'alignement et ainsi diminuer leur dissimilarité.
|
1 2 |
AGCGTACAT-GC A-CGCACGTCAC |
Ainsi dans l'alignement ci-dessus on ne compte plus que 5 différences. Il est tout à fait légitime de chercher à placer des lacunes au sein même des séquences puisque les mutations biologiques peuvent être non seulement des substitutions (remplacements de nucléotides) mais aussi des insertions ou des délétions (suppressions) de nucléotides.
Peut-on faire encore mieux ? Peut-on améliorer cet alignement pour diminuer encore le nombre de différences ? Juger de l'optimalité d'un alignement à l'œil nu est impossible. Il nous faut un algorithme permettant de le vérifier.
C'est là qu'entre en jeu l'algorithme de Needleman-Wunsch, un grand classique de la bioinformatique. Mais ses applications vont bien au-delà du domaine de la biologie. Par exemple, c'est ce même algorithme qu'on utilise pour calculer la distance d'édition entre tous les mots du dictionnaire et le mot que vous avez mal orthographié (donc absent du dictionnaire) et qui est souligné avec des zigzags rouges par votre logiciel de traitement de texte, afin de vous suggérer une petite liste de mots très similaires pour le remplacer.
Je ne rentrerai pas ici dans les détails complexes de l'alignement multiple de séquences (ou MSA pour Multiple Sequence Alignement) ni dans la jungle des logiciels permettant de produire de tels alignements. L'objectif de cet article est plutôt de vous expliquer le principe de base permettant d'aligner seulement deux séquences. En bonus je vous offre une implémentation de Needleman-Wunsch en annexe à la fin de l'article.

1. La distance d'édition
La distance d'édition est le nombre minimal d'opérations nécessaires pour transformer une chaîne de caractères en une autre. Les trois opérations autorisées sont l'insertion d'un caractère, la suppression (délétion) d'un caractère, et le remplacement (substitution) d'un caractère.
Quelle est par exemple la distance d'édition entre les mots SANG et ETANG ? Une manière simple de procéder serait de supprimer les 4 caractères de SANG puis d'insérer les 5 caractères de ETANG, ce qui fait 9 opérations. Ce n'est évidemment pas optimal.
Vous avez sans doute remarqué que les trois dernières lettres sont identiques. Il est donc possible de transformer le mot SANG en insérant un E au début puis en remplaçant le S par un T. Il n'est pas possible de faire plus court, la distance d'édition est par conséquent de 2 (remarquez que remplacer une lettre par une autre n'est pas équivalent à une délétion suivie d'une insertion car cela compterait comme deux opérations et non une seule).
En fait, toute suite d'opérations transformant une séquence en une autre correspond à un alignement particulier. Par exemple supprimer toutes les lettres de SANG et insérer toutes les lettres de ETANG correspond à cet alignement :
|
1 2 |
SANG----- ----ETANG |
Les deux opérations optimales, insérer le E et changer le S en T, correspondent à l'alignement suivant :
|
1 2 |
-SANG ETANG |
2. Dénombrer et représenter les alignements
Combien d'alignements différents existe-t-il ? La réponse à cette question s'appelle le nombre de Delannoy (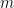
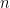

Ce qu'il faut en retenir c'est que ce nombre croît très rapidement. Pour notre tout petit cas de 4 et 5 lettres, cela donne déjà 681 alignements différents ! Et pour le couple de séquences de 11 nucléotides vu en introduction cela donne 45 046 719 alignements. Il est clair qu'on ne va pas tous les examiner un par un… Quoique ?
Bien qu'il ne soit pas envisageable de lister explicitement tous ces alignements différents, il est possible de les représenter comme des chemins dans une matrice en deux dimensions.
Les lettres de la première séquence sont utilisées comme indices des lignes et les lettres de la deuxième séquence sont utilisées comme indices des colonnes. On peut alors dessiner le chemin suivant où une flèche allant vers le bas indique qu'on supprime la lettre de la ligne suivante (puisqu'on parcourt les indices de la séquence SANG alors que l'indice de la séquence ETANG ne change pas), et où une flèche allant vers la droite indique qu'on insère la lettre de la colonne suivante (puisque l'indice de la séquence SANG ne change pas alors qu'on parcourt les indices de ETANG).
| E | T | A | N | G | ||
| ⭣ | ||||||
| S | ⭣ | |||||
| A | ⭣ | |||||
| N | ⭣ | |||||
| G | ⭢ | ⭢ | ⭢ | ⭢ | ⭢ | ⚑ |
Nous avons bien sûr vu précédemment que cet alignement n'était pas du tout optimal. Voici maintenant le chemin suivi par le meilleur alignement, la flèche en diagonal signifie qu'on réalise une substitution de la lettre de la ligne suivante par la lettre de la colonne suivante, ou bien que ces deux lettres sont identiques :
| E | T | A | N | G | ||
| ⭢ | ⭨ | |||||
| S | ⭨ | |||||
| A | ⭨ | |||||
| N | ⭨ | |||||
| G | ⚑ |
Les chemins représentés dans ces matrices doivent toujours relier la cellule tout en haut à gauche et la cellule tout en bas à droite.
3. Les paramètres de score
Dans ces matrices chaque mouvement réalisé a un coût. Ces coûts s'ajoutent lorsqu'on parcourt le chemin, et on obtient ainsi un score d'alignement lorsqu'on atteint la cellule d'arrivée. Trouver l'alignement optimal revient donc à trouver le chemin qui maximise ce score d'alignement. Lorsque les indels (insertions/délétions ou gaps) et les substitutions (mismatches) ont une pénalité de -1 et que les identités (matches) ont un coût de 0, alors le score d'alignement est égal à l'opposé de la distance d'édition. On utilise généralement ces paramètres dans les correcteurs d'orthographe, mais ils ne sont pas adaptés aux séquences d'ADN ou de protéines, où il est important d'attribuer une récompense positive aux identités.
Avec les paramètres match = 1 / mismatch = -1 / gap = -1, l'alignement suivant a un score d'alignement de 1 (-1, -1, +1, +1, +1) :
|
1 2 |
-SANG ETANG |
Et l'alignement suivant a un score d'alignement de 2 (+1, -1, +1, +1, -1, +1, +1, -1, +1, -1, -1, +1) :
|
1 2 |
AGCGTACAT-GC A-CGCACGTCAC |
On peut bien sûr utiliser des paramètres beaucoup plus complexes (par exemple pénaliser davantage les transversions que les transitions) mais cela ne change rien au principe de l'algorithme. Le score d'alignement peut être positif ou négatif, cela ne change rien non plus au fait qu'on cherche à le maximiser.
4. La notion d'homologie moléculaire
Mais pourquoi au juste est-il nécessaire de récompenser les identités pour les séquences biologiques ? Observons attentivement l'exemple suivant.
Alignement optimal avec les paramètres match = 0 / mismatch = -1 / gap = -1 :
|
1 2 |
TTTAAAAAGGGGGGTTT TTTCCCCCCAAAAATTT |
Alignement optimal avec les paramètres match = 1 / mismatch = -1 / gap = -1 :
|
1 2 |
TTT------AAAAAGGGGGGTTT TTTCCCCCCAAAAA------TTT |
Si on ne récompense pas les identités le premier alignement a un score de -11 à cause de 11 substitutions, alors que le deuxième a un score de -12 à cause des 12 indels, le premier est donc optimal. On voit pourtant que le deuxième a biologiquement plus de sens, puisqu'il aligne la sous-chaîne de 5 A. L'interprétation du premier alignement serait que cette sous-chaîne de 5 A est apparue indépendamment dans les deux séquences.
Au contraire l'interprétation du deuxième alignement serait que cette sous-chaîne de 5 A est homologue dans les deux séquences et donc qu'elle a été héritée d'un ancêtre commun. La similarité entre deux séquences est un indice de leur potentielle homologie qui doit être pris en compte lors de l'alignement. En attribuant une récompense de 1 aux identités le premier alignement a maintenant un score de -5 (à cause des 6 T identiques) et le deuxième alignement a un score de -1 (à cause des 6 T et des 5 A identiques), le deuxième est donc optimal.
Il existe plusieurs critères classiques (que nous ne détaillerons pas) permettant de formuler une hypothèse d'homologie entre deux caractères anatomiques, et ce sont les mêmes critères qu'on utilise en biologie moléculaire. Le premier d'entre eux est évidemment la similarité des structures. Plus deux caractères anatomiques sont similaires et moins il est probable qu'ils soient apparus indépendamment, cela est d'autant plus vrai que ces caractères sont complexes. En termes moléculaires, cela revient à juger comme plus probablement homologues les sous-chaînes qui sont davantage similaires, surtout si elles sont longues. Le fait de récompenser les identités dans le calcul du score reflète bien ce critère intuitivement.
Il arrive cependant qu'une structure anatomique soit tellement modifiée au cours de l'évolution que la similarité ne soit plus un critère suffisant pour reconnaître cette homologie, par exemple une aile de chauve-souris et une nageoire de dauphin qui sont tous les deux des membres chiridiens homologues. Dans de tels cas c'est le critère de position qui peut nous venir en aide : deux structures homologues devraient être positionnées de la même façon par rapport aux structures voisines, elles-mêmes déjà reconnues comme homologues (ce critère a été formulé par Étienne Geoffroy Saint-Hilaire sous le nom de "principe des connexions").
Ainsi l'aile de la chauve-souris et la nageoire du dauphin s'articulent sur le même os, la scapula (omoplate), avec les mêmes muscles, et reçoivent les mêmes nerfs et les mêmes artères. En biologie moléculaire, si deux sous-chaînes dissimilaires dans différentes séquences sont encadrées par des sous-chaînes homologues, alors elles pourraient être considérées comme homologues elles aussi. Observons ce dernier exemple pour comment ce critère s'applique concrètement aux séquences biologiques.
Alignement optimal avec les paramètres match = 0 / mismatch = -1 / gap = -1 :
|
1 2 |
CGTGACTTT-ATTAAATTTAGTTGTC CGTTTTTTTGGGTTTTTTTCG-AGTC |
Alignement optimal avec les paramètres match = 1 / mismatch = -1 / gap = -1 :
|
1 2 |
CGTGACTTTATTAAATTTAGTT---GTC CGT---TTTTTTGGGTTTTTTTCGAGTC |
On voit que la récompense pour les identités nous permet de reconnaître comme homologues les 3 positions centrales occupées par AAA et GGG dans les deux séquences alors qu'il n'y a aucune identité entre ces deux sous-chaînes ! Cela est dû au fait que cette récompense permet la reconnaissance de l'homologie des deux sous-chaînes qui flanquent ces 3 positions centrales : à gauche TTTATT dans la première séquence et TTTTTT dans la deuxième séquence (une seule différence sur 6 positions), à droite TTTAGTT dans la première séquence et TTTTTTT (deux différences sur 7 positions). C'est donc parce que les sous-chaînes flanquantes ont été reconnues comme homologues que les sous-chaînes centrales ont été alignées.
5. La relation de récurrence de Needleman-Wunsch
L'algorithme de Needleman-Wunsch repose sur le principe de la programmation dynamique pour résoudre le problème de l'alignement global optimal (le problème de l'alignement local utilise une variante, l'algorithme de Smith-Waterman, que nous n'aborderons pas ici). L'idée est de décomposer le problème en sous-problèmes plus simples. Ainsi, au lieu de tenter d'aligner directement les deux séquences de longueurs 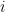



Tout d'abord, l'alignement optimal et le score maximal correspondant sont évidents si l'une des deux chaînes est vide (0 caractère). Le seul moyen d'aligner les 

| E | T | A | N | G | ||
| 0 | -1 | -2 | -3 | -4 | -5 | |
| S | -1 | |||||
| A | -2 | |||||
| N | -3 | |||||
| G | -4 |
On veut maintenant déterminer 
Admettons que l'alignement optimal correspondant est connu et donc que
- l'insertion du dernier caractère
de la deuxième séquence ;
- la délétion du dernier caractère
de la première séquence ;
- ou bien la "substitution" du dernier caractère
Ces trois opérations correspondent à l'une des trois flèches possibles que nous avons vu précédemment (cf. Section 2).
Disons qu'on se trouve dans le premier cas de figure et que la dernière opération est l'insertion du caractère 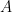


Par un raisonnement similaire on peut démontrer que si 



Dans un cas réel on ne connaît pas la dernière opération, mais on veut que

Grâce à l'initialisation de la matrice, on connaît déjà toutes les valeurs de 






| E | T | A | N | G | ||
| 0 | -1 | -2 | -3 | -4 | -5 | |
| S | -1 | -1 | -2 | -3 | -4 | -5 |
| A | -2 | -2 | -2 | -1 | -2 | -3 |
| N | -3 | -3 | -3 | -2 | 0 | -1 |
| G | -4 | -4 | -4 | -3 | -1 | 1 |
6. Le retour sur trace (backtracking)
Une fois que la matrice des scores 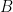
Par exemple, si parmi les trois scores possibles, 



Si c'est le second score qui est le plus grand on inscrit une flèche vers la gauche ⭠ indiquant que le score maximal précédent la dernière opération se trouve dans la cellule juste à gauche, c'est-à-dire 

En cas d'égalité entre plusieurs de ces scores cela signifie que l'alignement optimal n'est pas unique, plusieurs alternatives ont le même score maximal. On peut alors soit décider d'inscrire plusieurs flèches dans la même cellule, si on souhaite énumérer exhaustivement tous les alignements optimaux, ou bien arbitrairement une seule, si on ne souhaite obtenir qu'un seul des alignements optimaux. Généralement c'est la seconde option est choisie, par souci d'efficacité des calculs.
| E | T | A | N | G | ||
| ⚑ | ⭠ | ⭠ | ⭠ | ⭠ | ⭠ | |
| S | ⭡ | ⭦ | ⭦ | ⭦ | ⭦ | ⭦ |
| A | ⭡ | ⭦ | ⭦ | ⭦ | ⭠ | ⭠ |
| N | ⭡ | ⭦ | ⭦ | ⭡ | ⭦ | ⭠ |
| G | ⭡ | ⭦ | ⭦ | ⭡ | ⭡ | ⭦ |
L'idée est ensuite de suivre les flèches qui mènent de la cellule 


- Une flèche en diagonale, alors on copie dans l'alignement final le caractère
- Une flèche vers la gauche, alors on copie un caractère de gap "-" dans la première séquence alignée et le caractère
- Une flèche vers le haut, alors on copie le caractère
On continue ainsi à rebours jusqu'à ce que 

Dans notre exemple avec SANG et ETANG on construit ainsi successivement les alignements suivants :
⭦ , c'est une substitution (match) du caractère 3 de la première séquence par le caractère 4 de la deuxième séquence :
|
1 2 |
G G |
⭦ , c'est une substitution (match) du caractère 2 de la première séquence par le caractère 3 de la deuxième séquence :
|
1 2 |
NG NG |
⭦ , c'est une substitution (match) du caractère 1 de la première séquence par le caractère 2 de la deuxième séquence :
|
1 2 |
ANG ANG |
⭦ , c'est une substitution (mismatch) du caractère 0 de la première séquence par le caractère 1 de la deuxième séquence :
|
1 2 |
SANG TANG |
⭠ , c'est une insertion du caractère 0 de la deuxième séquence (ce qui correspond donc à un gap dans la première séquence) :
|
1 2 |
-SANG ETANG |
Et voilà maintenant vous savez comment effectuer un alignement global de deux séquences !
Conclusion
L'algorithme de Needleman-Wunsch a été plusieurs fois redécouvert ou réinventé dans différents domaines. Le nom de l'algorithme vient bien sûr de la publication de 1970 par Saul Needleman et Christian Wunsch, mais ils ne sont ni les premiers (Vintsyuk, 1968) ni les derniers à avoir publié cette idée (Wagner & Fischer, 1974). Je n'ai d'ailleurs pas présenté la version originale de Needleman et Wunsch, mais une version modernisée telle qu'elle est aujourd'hui généralement enseignée aux débutants.
Diverses améliorations de cet algorithme sont possibles, que ce soit pour le rendre plus efficace en termes de complexité spatiale ou temporelle, ou bien en termes d'adéquation des paramètres à la réalité biologique.
Par exemple en ce qui concerne les possibles économies de stockage, on peut remarquer que la matrice des scores
En ce qui concerne l'adéquation aux mécanismes biologiques, il est classique de pointer du doigt l'irréalisme de la modélisation des pénalités des gaps par des fonctions linéaires. Autrement dit un gap de longueur 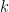


On préfère presque toujours les fonctions affines qui associent à un gap une forte pénalité d'ouverture 


Enfin soulignons que l'algorithme tel que je l'ai présenté ici n'est valable que pour l'alignement global de deux séquences seulement. Si on souhaite aligner davantage de séquences en étendant naïvement la logique de Needleman-Wunsch, alors il faudrait remplir une matrice des scores cubique pour 3 séquences, puis un hypercube à 4 dimensions pour 4 séquences, etc. Ainsi la complexité temporelle de ce genre d'algorithme serait 
Références
- Torres, A., Cabada, A., & Nieto, J. J. (2003). An exact formula for the number of alignments between two DNA sequences. DNA Sequence, 14(6), 427-430.
- Gusfield, D. (1997). Algorithms on Strings, Trees, and Sequences : Computer Science and Computational Biology. Cambridge : Cambridge University Press.
- Wagner, R. A., & Fischer, M. J. (1974). The string-to-string correction problem. Journal of the ACM (JACM), 21(1), 168-173.
- Needleman, S. B., & Wunsch, C. D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of molecular biology, 48(3), 443-453.
- Vintsyuk, T. K. (1968). Speech discrimination by dynamic programming. Cybernetics, 4(1), 52-57.
Annexe : implémentation de Needleman-Wunsch en Python
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 |
def needleman_wunsch(seq1, seq2, match, mismatch, gap): # Définition des constantes pour les directions dans la matrice des traces GAUCHE = 0 # Insertion (déplacement vers la gauche) HAUT = 1 # Délétion (déplacement vers le haut) DIAGONALE = 2 # Substitution ou match (déplacement en diagonale) # Caractère utilisé pour représenter un gap GAP_CHAR = "-" # Longueurs des séquences m = len(seq1) n = len(seq2) # Initialisation de la matrice des scores (S) et de la matrice des traces (B) S = [[0] * (n + 1) for _ in range(m + 1)] B = [[None] * (n + 1) for _ in range(m + 1)] # Initialisation de la première colonne et de la première ligne for i in range(1, m + 1): S[i][0] = i * gap B[i][0] = HAUT for j in range(1, n + 1): S[0][j] = j * gap B[0][j] = GAUCHE # Remplissage de la matrice S et enregistrement du choix dans B for i in range(1, m + 1): for j in range(1, n + 1): if seq1[i - 1] == seq2[j - 1]: score_diag = S[i - 1][j - 1] + match else : score_diag = S[i - 1][j - 1] + mismatch score_haut = S[i - 1][j] + gap score_gauche = S[i][j - 1] + gap # Choix de la meilleure option if score_diag >= score_haut and score_diag >= score_gauche : S[i][j] = score_diag B[i][j] = DIAGONALE elif score_gauche >= score_haut : S[i][j] = score_gauche B[i][j] = GAUCHE else : S[i][j] = score_haut B[i][j] = HAUT # Backtracking pour reconstituer l'alignement optimal align1 = "" align2 = "" i = m j = n while i > 0 or j > 0 : if B[i][j] == DIAGONALE : align1 = seq1[i - 1] + align1 align2 = seq2[j - 1] + align2 i -= 1 j -= 1 elif B[i][j] == GAUCHE : align1 = GAP_CHAR + align1 align2 = seq2[j - 1] + align2 j -= 1 else : align1 = seq1[i - 1] + align1 align2 = GAP_CHAR + align2 i -= 1 return S[m][n], align1, align2 # Exemple d'utilisation simple seq1 = "CGGTTCGAAC" seq2 = "CAGCCATTAC" score, align1, align2 = needleman_wunsch(seq1, seq2, 1, -1, -1) print("Score optimal :", score) print("Séquence 1 :", align1) print("Séquence 2 :", align2) |
Merci à Léopold Carron et ZaZo0o pour la relecture.
Laisser un commentaire