

Étiquette : BLAST
-
Automatiser la récupération de données biologiques : version avancée
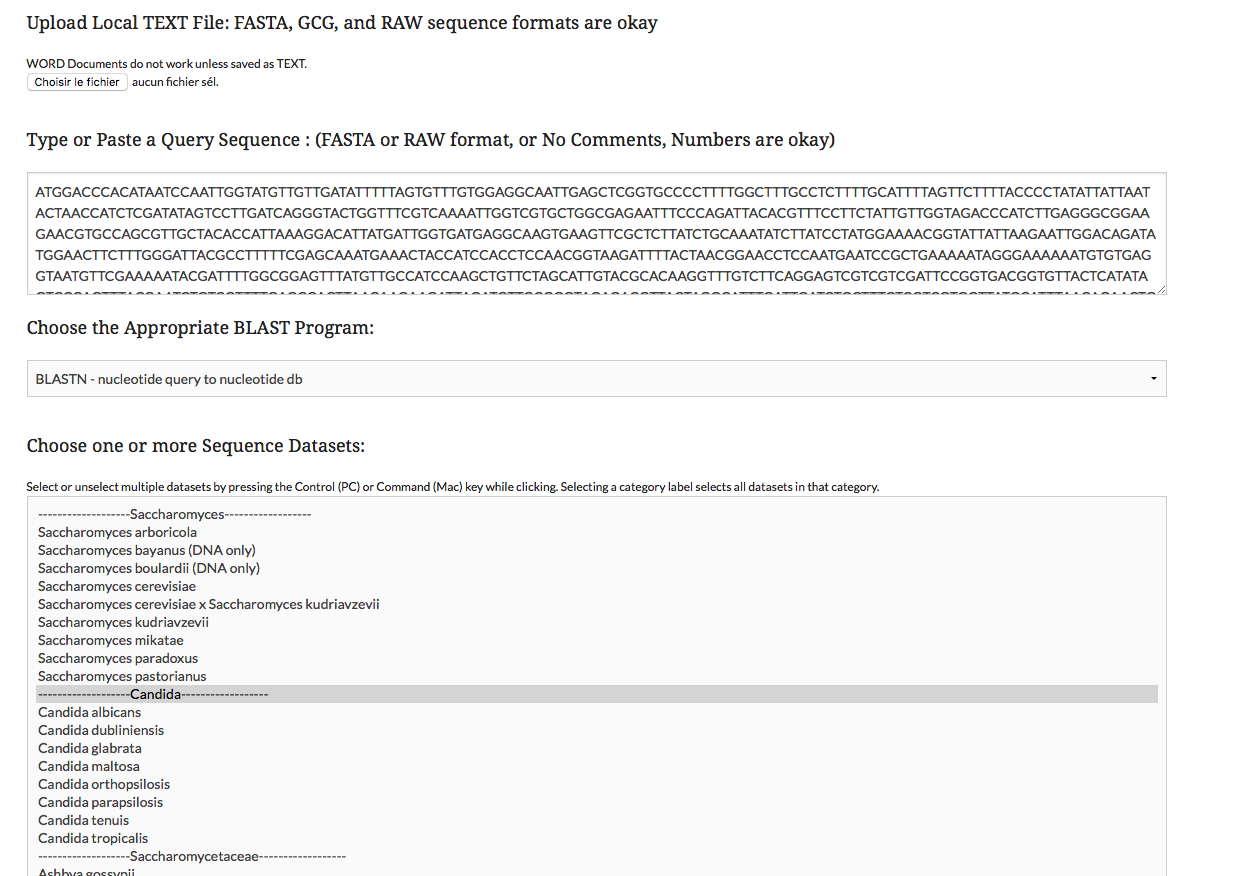
Dans cette version avancée, nous allons nous intéresser à une autre manière de récupérer des données depuis un site internet : via l'utilisation de formulaires. Les formulaires, ce sont ces pages avec plein de champs à remplir et que vous soumettez ensuite pour vous créer un compte, contacter votre hotline ou que sais-je. Leur utilisation permet…
-
Questions à… Laurent Mouchard

Avec un peu beaucoup de retard, retrouvez la retranscription de la TOBi organisée par JeBiF en mai 2016 avec Laurent Mouchard, maître de conférence à l'Université de Rouen et modérateur de la liste bioinfo. Nous le remercions d'être venu nous raconter la petite histoire de la bioinformatique ! . Laurent Mouchard : Je suis vieux… J'ai donc eu le temps…
-
BLAST en pratique

Cet article a pour but de vous montrer une application pratique de BLAST, le fameux programme d'alignement de séquences détenant un record de citations, avec certains problèmes qu'on peut rencontrer et ce qu'on peut tirer de son résultat. BLAST a au moins deux usages typiques en génomique : Pour les initiés, le premier point est à…
-
Soirée BED & FASTA !

Après la petite histoire de l’analyse des séquences d’ADN, voici un tutoriel pour apprendre quelques trucs et astuces dans ce domaine. Biologiste en mal de connaissances de programmation ou pro de R, vous trouverez ici de quoi vous amuser avec un fichier Fasta ou un Bed. Nous allons voir comment faire un alignement multiple de…
-
Transformer une sortie de Blast xml en format GenBank
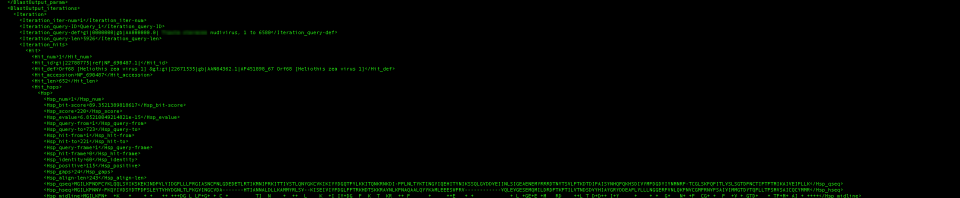
Bonjour, et bienvenu dans mon premier article concernant mon script Perl Blast2Gb.pl. Ce dernier illustre parfaitement ce qu'on peut faire facilement et rapidement avec un script Perl : du traitement de texte. Cet outil permet de faciliter la vie et de transformer une tâche laborieuse en un traitement rapide et efficace. En effet, qui ne s'est…