

Étiquette : séquençage
-
Comment l'étude des intensités d'expression de gène a évolué avec les technologies de transcriptomique ?
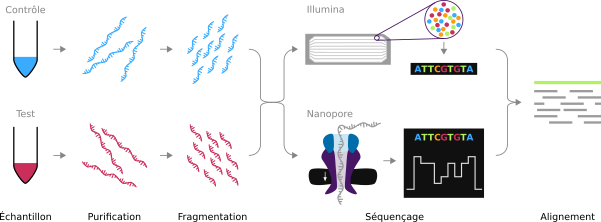
Cet article est en partie basé sur mon introduction de thèse [0], reformatée pour convenir au format du blog et agrémentée des nouvelles informations pour ne pas être un simple catalogue de connaissances. Qu'il s'agisse du génome entier, du génome de tissus ou du génome de cellules spécifiques, l'étude de la quantité de transcrits produits,…
-
Chronique d'une soumission de données à GEO

Je vais vous raconter étape par étape ma soumission de données de séquençage à la base de données de génomique GEO (Gene Expression Omnibus) d'un projet en cours de finition. Sommaire GEO, Qu'est-ce que c'est ? Lorsque l'on veut publier les résultats d'une étude comprenant du séquençage haut débit, nous devons publier les données brutes et…
-
R : représenter des genomic tracks avec Gviz
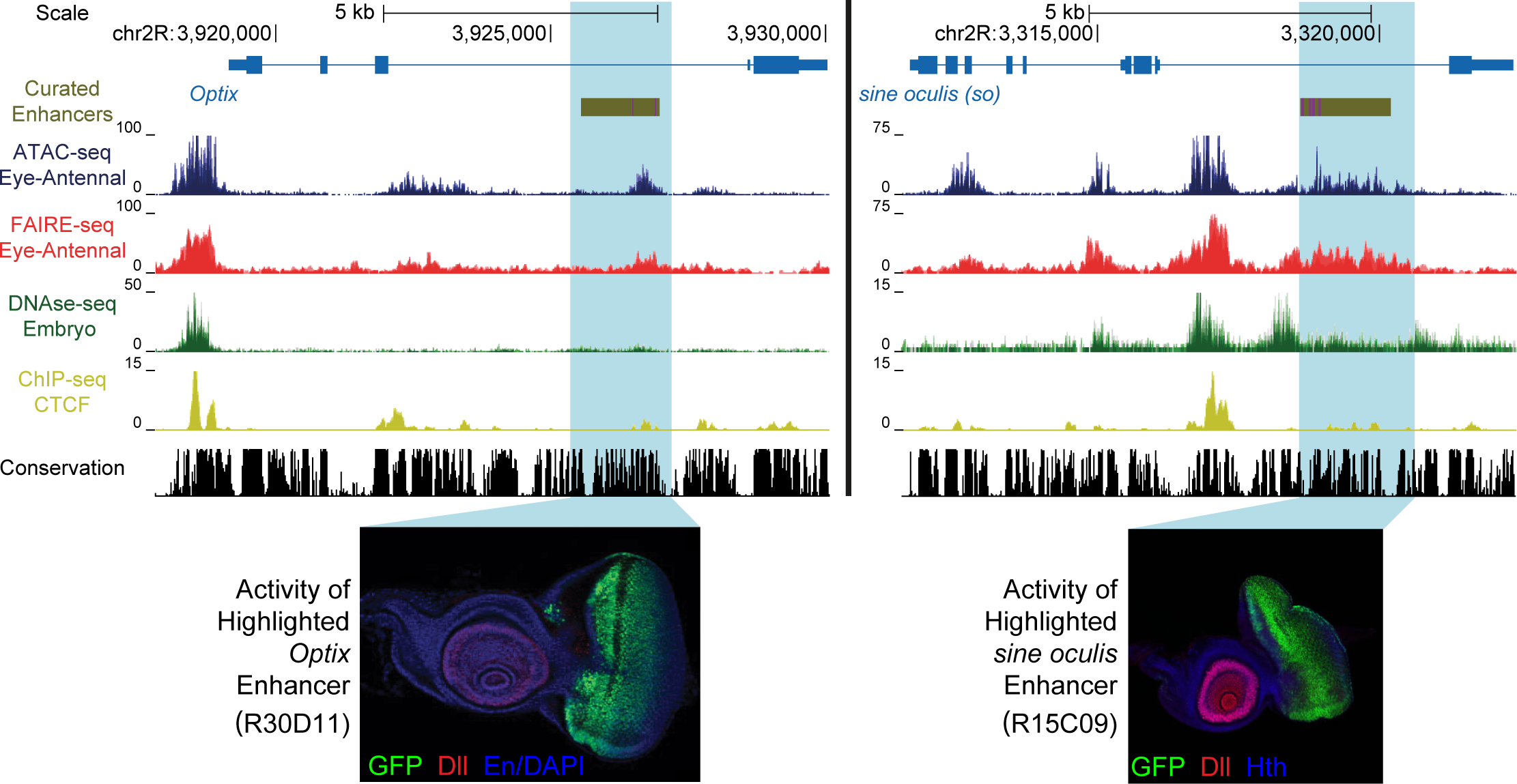
Si vous analysez des données d'épigénomique telles que de l'ATAC-seq ou des ChIP-seq, vous souhaitez sûrement pouvoir représenter des exemples de pics sous forme de genomic tracks comme on en voit souvent dans les publications. Lorsque l'on inspecte ses données de coverage (ou couverture), on charge généralement le fichier Bam ou le fichier BigWig dans…
-
Pourquoi certains fichiers FASTQ finissent par 001 ?

Sur le chan IRC du blog, un de nos membres se demandait pourquoi les noms de fichiers FASTQ devait finir par _001.fastq sur la plateforme de cloud computing d'Illumina BaseSpace. Mais avant de répondre à cette question pressante, repartons du début. Les fichiers FASTQ En cette période de domination du séquençage haut débit de l'ADN,…
-
1… 2… 3… 4C ! Ou comment capturer l'état de la chromatine.
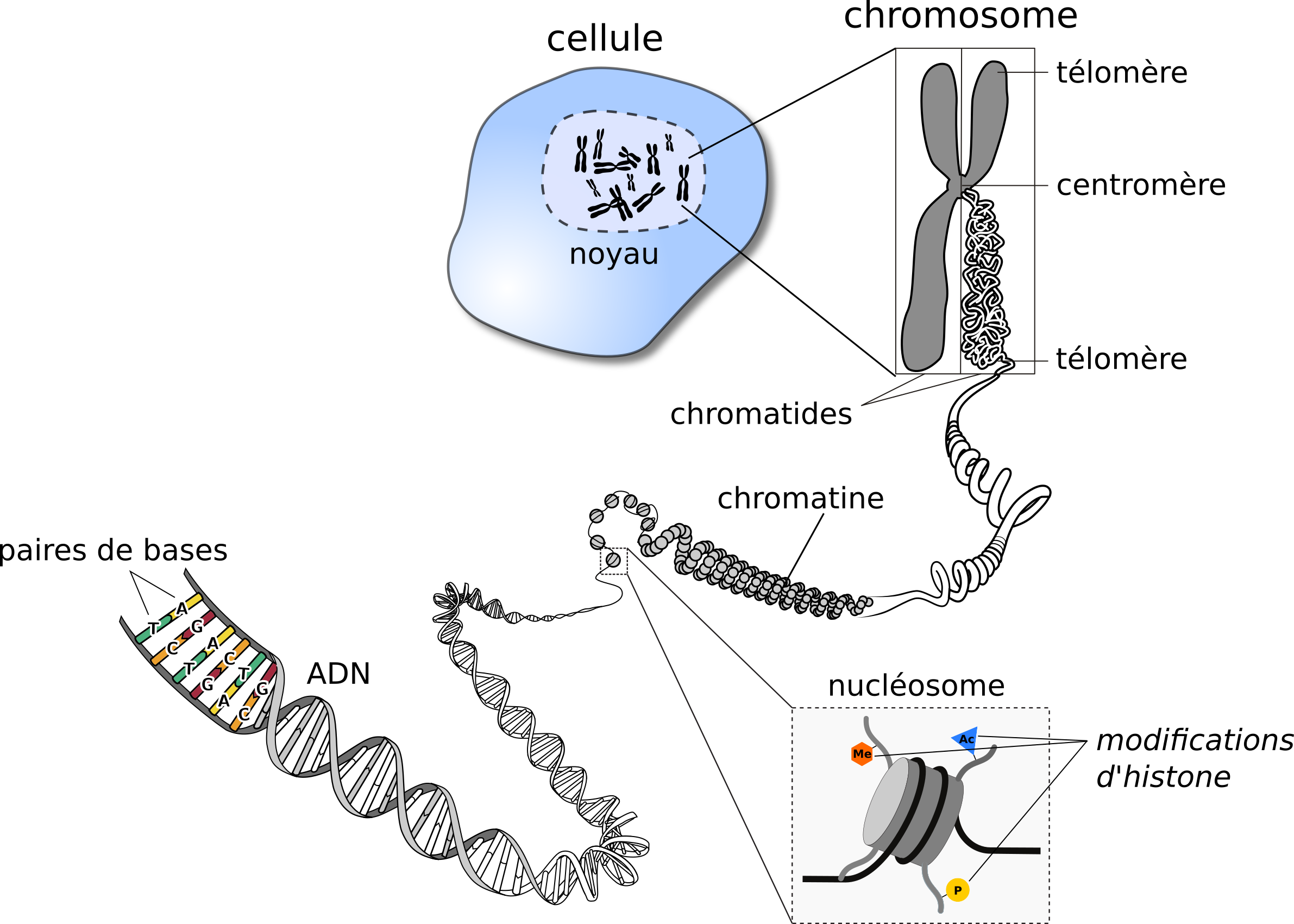
Une cellule eucaryote comporte un noyau qui contient l’information génétique portée par les chromosomes, eux même composés d’ADN. Chez l'Homme, l'ADN des chromosomes mis bouts à bouts mesure 1,9 mètre de long. Or, tout ce matériel génétique doit tenir dans le noyau des cellules, qui lui mesure 5 à 7 micromètres de diamètre (en moyenne…
-
Génomique des paysages

Introduction « Génomique des paysages » cela sonne comme le titre d’une œuvre d’Eduardo Kac. Ce nom un peu post-moderne désigne en fait une discipline scientifique qui a connu une expansion fulgurante au cours de la dernière décennie. Les enjeux environnementaux et de conversation actuels ont rendus pressante la nécessité de mieux comprendre et décrire les espèces…
-
La bio-informatique au service de l’antibiorésistance
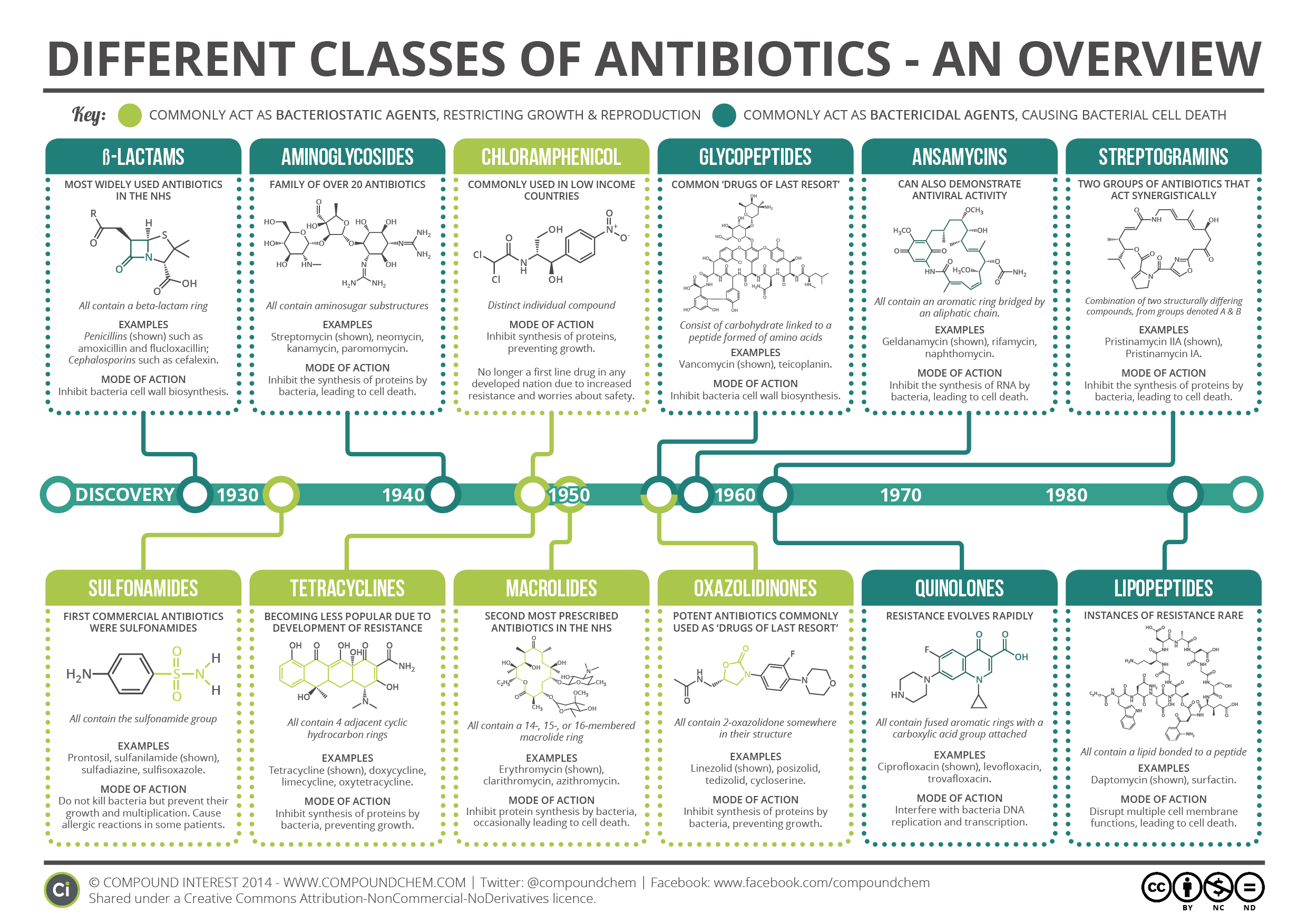
En ces temps hivernaux, virus et infections bactériennes sont de retour (pour vous jouer un mauvais tour…). Les campagnes de prévention et de soin sont donc de sortie, et parmi elles la célèbre : "Les antibiotiques, c'est pas automatique". Si ce slogan est bien rentré dans la tête des gens, la raison pour laquelle il a…
-
Traquer les régions ouvertes de l'ADN avec l'ATAC-seq
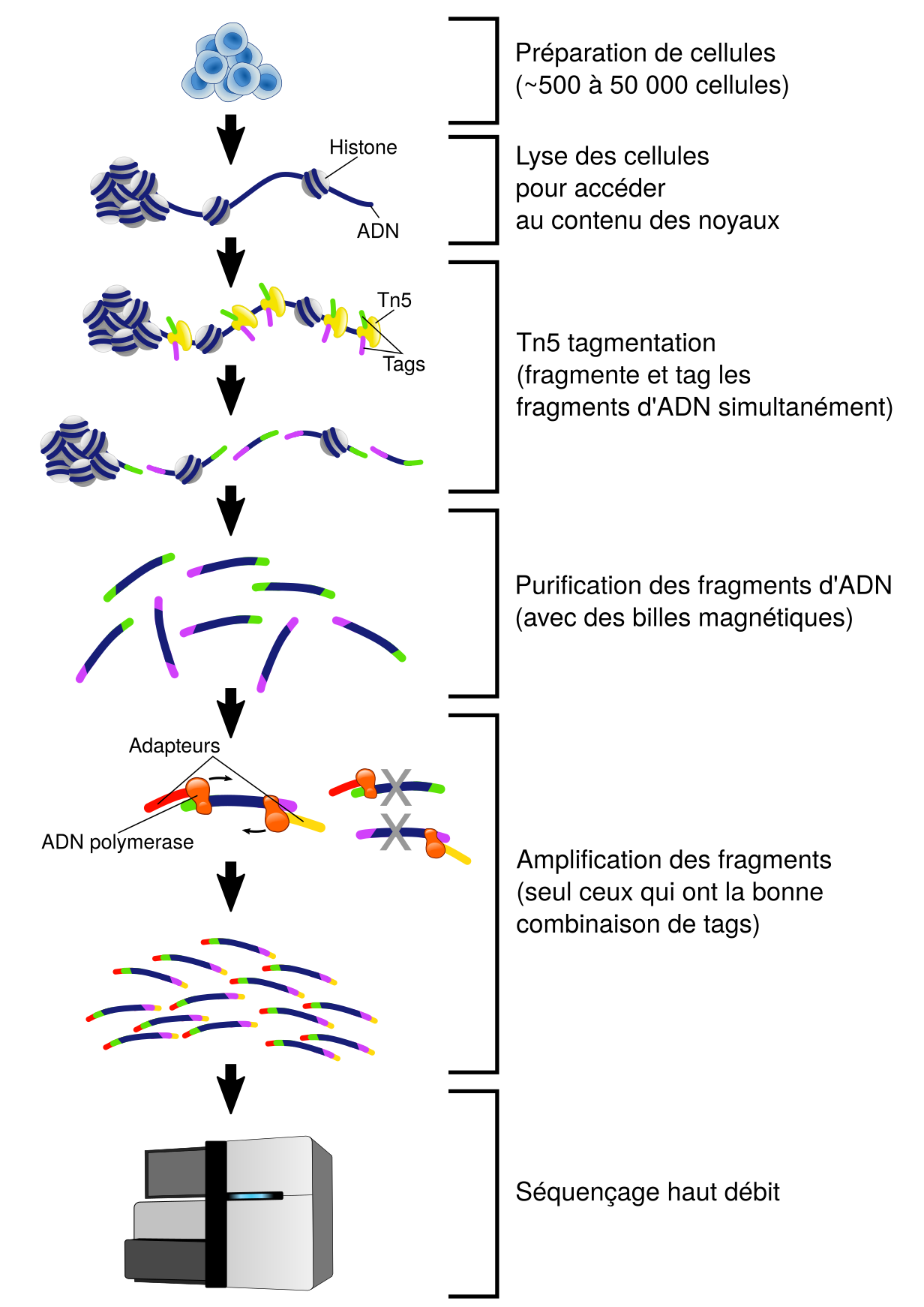
L'étude de la régulation de l'expression des gènes est une discipline complexe qui recoupe des données provenant de divers types d'expériences. Dans un précédent article, nous avions vu trois techniques de biologie moléculaire couplées à du séquençage haut débit classiquement employées pour mettre en évidence les régions accessibles de l'ADN, et donc potentiellement des régions…