

Étiquette : NGS
-
Chronique d'une soumission de données à GEO

Je vais vous raconter étape par étape ma soumission de données de séquençage à la base de données de génomique GEO (Gene Expression Omnibus) d'un projet en cours de finition. Sommaire GEO, Qu'est-ce que c'est ? Lorsque l'on veut publier les résultats d'une étude comprenant du séquençage haut débit, nous devons publier les données brutes et…
-
ViLoVar : un outil pour la visualisation de variations génétiques
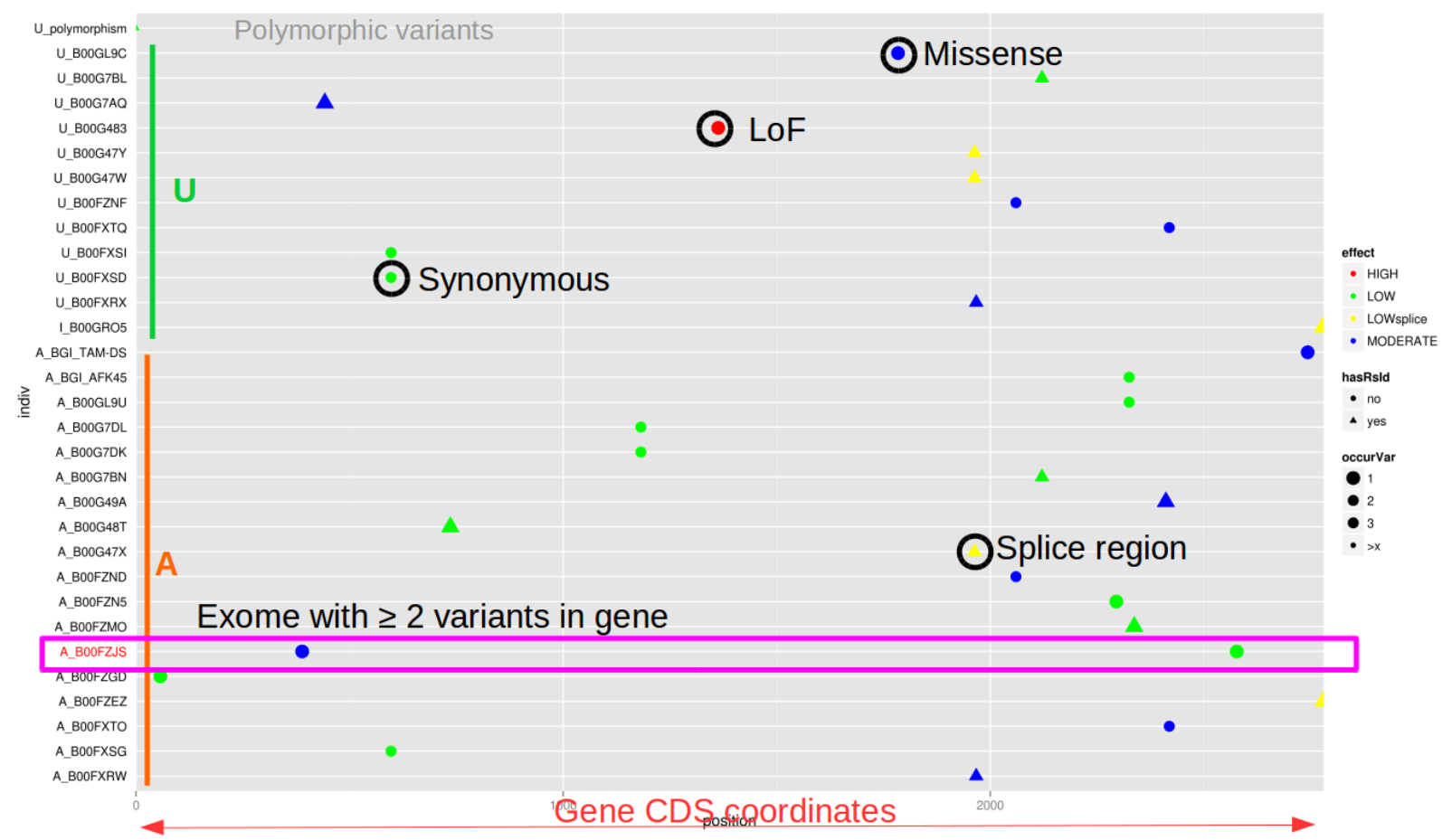
Pour mon premier article, je vais vous présenter un outil que j'ai développé lorsque je travaillais sur le projet "Myocapture"; un projet national de séquençage d'exomes qui portait sur les myopathies (https://www.afm-telethon.fr/myopathie-congenitale-6675). Ce projet visait à trouver de nouvelles mutations responsables de ces maladies rares. Il a également permis d'identifier de nouveaux gènes impliqués dans…
-
Vous ne savez pas comment analyser vos données Hi-C ? Exemple d'utilisation de HiC-pro
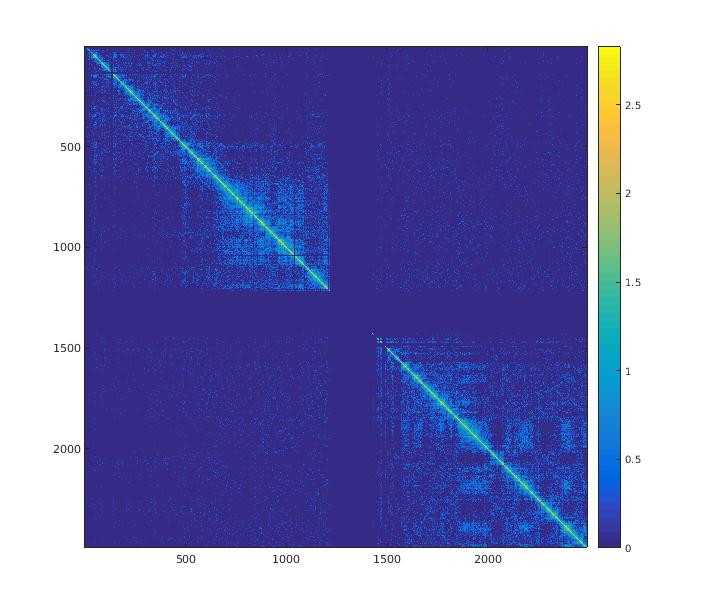
Partir de quelconques données de séquençage haut débit brutes pour arriver à une analyse complète demande au mieux, une certaine pratique de ces technologies. Dans bien des cas, on va alors mettre en place un pipeline reposant sur tout un tas d'outils. Il faudra probablement des heures pour comprendre les paramètres de chacun d’entre eux…
-
Introduction à l'analyse des SNPs
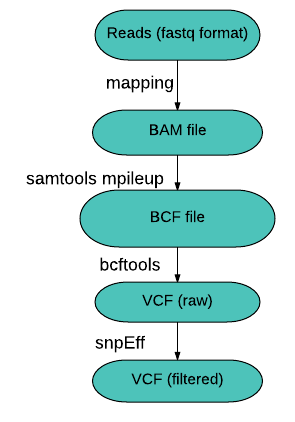
Introduction Un SNP (Single Nucleotid Polymorphism) est la mutation d'un seul nucléotide à une position donnée, entre individus d'une même population. Ces substitutions ponctuelles permettent d'identifier des sous-populations. Vous avez sûrement étudié en cours l'exemple de la drépanocytose, une maladie causée par la mutation ponctuelle d'un gène. L'origine de cette maladie génétique peut être observée en…
-
Hi-C : Quelques bases
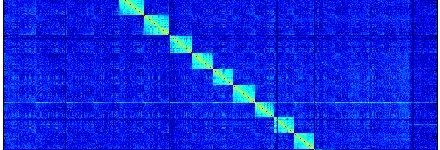
Aujourd'hui on va découvrir ensemble une des petites dernières dans la famille des techniques hauts débits : le High Chromosome Contact map (Hi C)[1] . Revenons sur quelques bases : un gène ne pourra être exprimé que si l'ADN qui le code est déplié. Par conséquent les régions dans lesquelles les gènes ne sont pas exprimés sont…
-
JOBIM2013 : le bilan

Ça y est, les JOBIM 2013 ont eu lieu. Cela se passait à Toulouse au Centre des Congrès Pierre Baudis. Ce fut une semaine bien chargée et bioinformatiquement très intéressante. Les sessions plénières ainsi que les parallèles ont rencontré un certain succès. Cette année les conférenciers invités étaient au nombre de six : Simon Anders (EMBL,…
-
Galaxy : Bien plus qu'un gestionnaire de workflows

Qu'est-ce que Galaxy ? Galaxy est une application web écrite en Python destinée à faciliter la manipulation et l'analyse des données, dans le cadre de la recherche biomédicale. Elle permet d'utiliser des logiciels habituellement exécutés en ligne de commande de manière graphique, grâce à un système de plugins (« outils ») en XML et de templates Mako. Ces…
-
Revue de presse : Le printemps, les oiseaux et … la biblio

Je dois l'avouer : ce mois est un cauchemar. Il y avait une tonne de choses passionnantes ! Alors, le choix d'en laisser certains en dehors m'a causé des nuits blanches… Je plaisante. J'en ai choisi donc quelques-unes espérant que le mélange gourmand et croquant fasse de l'ombre aux émissions culinaires de M6 (clin d'oeil aux Guignols).…