

Catégorie : Astuce
-
Trouver un emploi/une thèse en bioinformatique : quelques pistes [maj]
![Trouver un emploi/une thèse en bioinformatique : quelques pistes [maj]](https://bioinfo-fr.net/wp-content/uploads/2018/11/job-search.jpg)
Comme le disait Estel en 2012, trouver un job en bioinfo n'est pas évident. Contrairement à certains métiers qui concentrent l'entièreté des offres d'emploi de leur pays en une seule plateforme, les emplois de bioinfo sont distribuées aléatoirement entre des dizaines de sites d'annonces plus ou moins spécifiques à la bioinformatique. C'est pourquoi l'article d'Estel…
-
Customiser matplotlib (faire son matplotlibrc)

Suite à une mésaventure liée à matplotlib sur le chan IRC #bioinfo-fr (mésaventure suite aux fameuses erreurs de display ; si vous voulez tout savoir : si on configure mal son matplotlib on peut générer des erreurs qui font qu'on obtient des images vides… voir la partie sur le backend plus tard :o), j'ai parlé de la…
-
Maîtrisez le cache de Rmarkdown !

Pour des raisons de reproduction de la science, il est important de conserver une trace de tout ce que l'on fait sur son ordinateur. Pour cela, faire des rapports est la meilleure manière que je connaisse qui permette d'inclure le code et les résultats d'une analyse. Pour faire ça bien avec R, on a déjà…
-
Représenter rapidement une ACP avec R et ggplot2
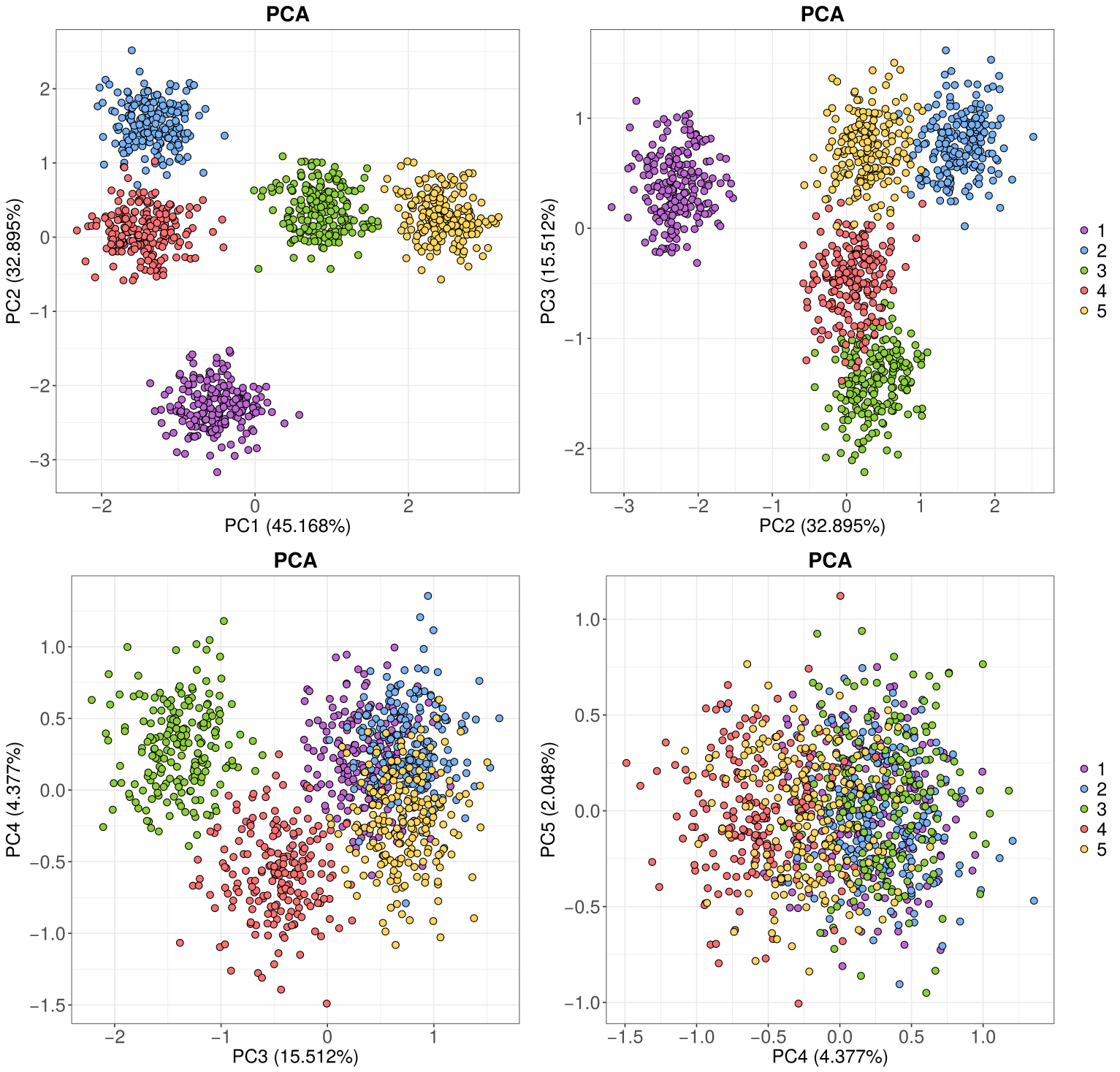
Je ne sais pas pour vous, mais moi, à chaque fois que j'assiste à une réunion de labo, il y a quasi systématiquement un graphique d'ACP pour montrer les données. Et à chaque fois, il s'agit d'un graphique de base, généré avec R, avec la fonction plot(), des couleurs qui piquent les yeux et des…
-
Comment fixer les problèmes de déploiement et de durabilité des outils en bioinformatique ? Indice : conda !
La diversité des questions que se posent nos amis biologistes entraîne une diversité des données : génomiques, images, etc. De plus, ces données sont générées à des vitesses folles. Pour manipuler les données et extraire les informations utiles, des solutions et outils bioinformatiques sont nécessaires. De nombreux outils existent déjà pour répondre à de nombreuses questions.…
-
S'outiller et s'organiser pour mieux travailler

TL;DR La reproductibilité, c’est la vie (dans le monde scientifique) ! Tout résultat doit pouvoir être reproduit. La technologie permet de faciliter la recherche de reproductibilité. Les cahiers de laboratoire papiers ne sont plus du tout adaptés à la recherche actuelle et au besoin de reproductibilité. Je préconise donc d’utiliser git et GitHub, de bien…
-
C'est l'enfeR.

Certains bio-informaticiens ne jurent que par R (j'en fais partie). Je suis amoureux de sa simplicité (sic), son élégance (re-sic), sa documentation et ses innombrables packages tous plus utiles les uns que les autres. Et surtout c'est le seul langage que je maîtrise un peu convenablement, alors forcément je trouve tous les autres langages nuls,…
-
Snakemake pour les nuls (ou comment créer un pipeline facilement ?)

Bonjour à tous, et bienvenue dans le premier épisode d'une (longue ?) série de prise en main de l'outil dédié au pipeline : Snakemake. Si vous ne connaissez pas encore cet outil, c'est que vous êtes sûrement passés à côté de cet article écrit par Nisaea. Alors, quel sera les bénéfices de retranscrire vos pipelines déjà…