

Étiquette : R
-
R : Convertir des Ensembl IDs en symboles de gènes

Lorsque l'on traite des données de RNA-seq, il arrive très souvent de se retrouver avec une matrice de quantification de l'expression des gènes (un tableau avec le nombre de reads par gène) dont le nom des gènes est représenté par leur identifiant (ou ID) de chez Ensembl (ex. : "ENSG00000128573") et non par leurs symboles…
-
R : représenter des genomic tracks avec Gviz
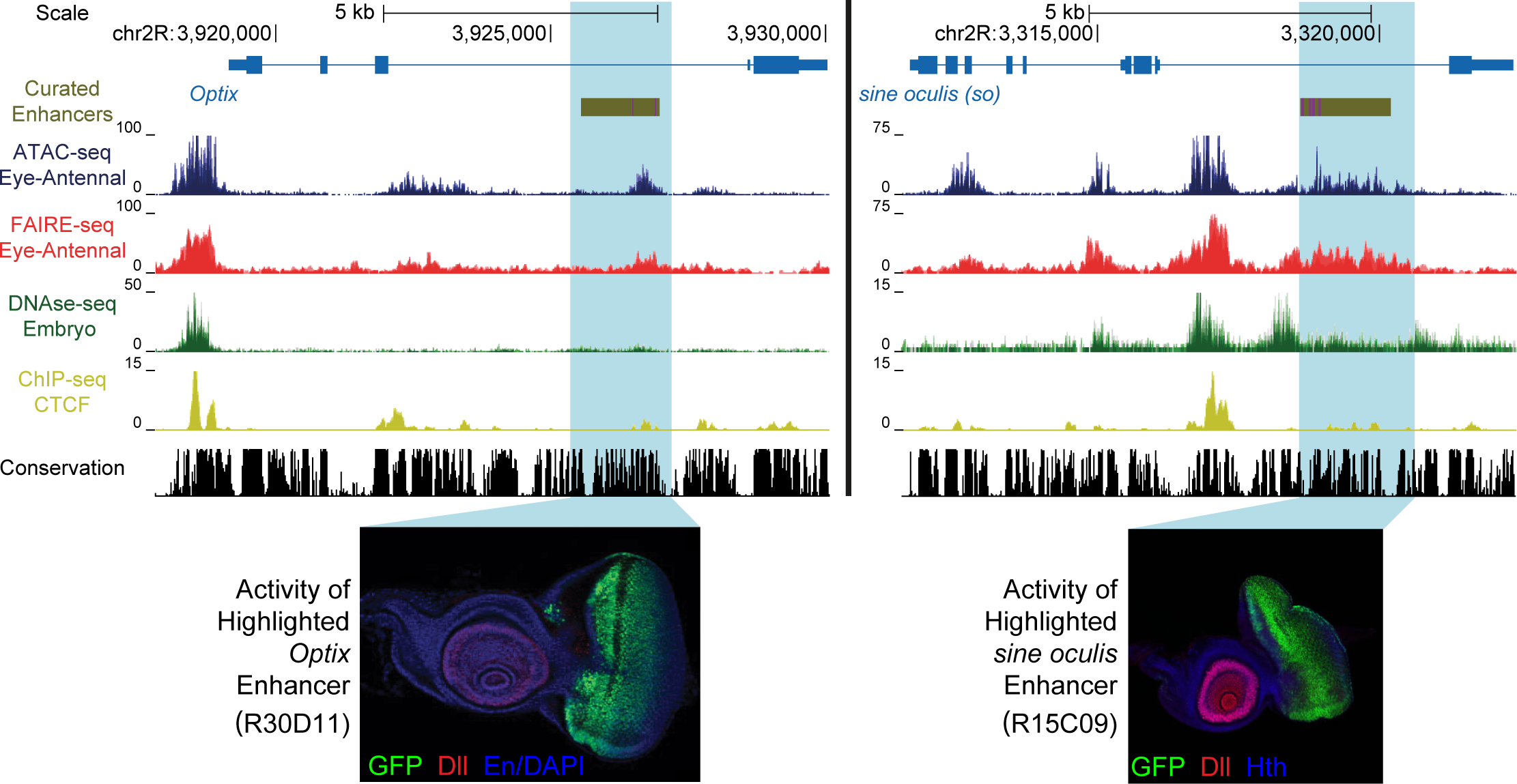
Si vous analysez des données d'épigénomique telles que de l'ATAC-seq ou des ChIP-seq, vous souhaitez sûrement pouvoir représenter des exemples de pics sous forme de genomic tracks comme on en voit souvent dans les publications. Lorsque l'on inspecte ses données de coverage (ou couverture), on charge généralement le fichier Bam ou le fichier BigWig dans…
-
Fréquences des dinucléotides dans le génome d'organismes modèles
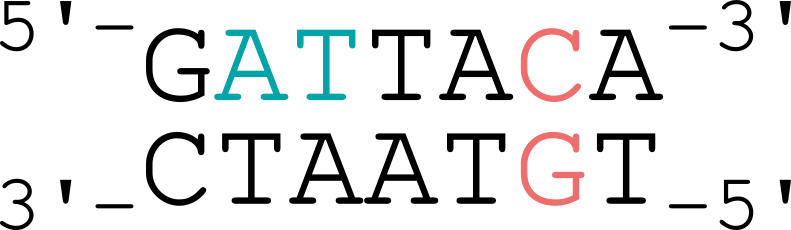
L'analyse de séquences est au cœur de nombreux domaines de la bio-informatique. Le billet du jour s'intéressera aux séquences ADN, en se proposant de compter la fréquence en dinucléotides dans quelques génomes d'organismes modèles (avec une petite arrière-pensée derrière la tête). Qu'est-ce qu'un dinucléotide ? L'ADN double brins est classiquement structuré sous forme de double hélice,…
-
Manipulation d'intervalles génomiques dans R
Introduction Nous avons abordé, dans le précédent article de cette série, les bases de la manipulation d'intervalles dans R. Ce deuxième article a pour objectif de montrer comment manipuler des intervalles génomiques. Au niveau le plus basique, un intervalle est défini par deux nombres entiers positifs délimitant son début et sa fin. Nous allons pouvoir…
-
Introduction à la manipulation d'intervalles dans R
Introduction "Quelle est la profondeur de ce séquençage ?" "Quelle proportion de SNPs se situent dans des exons ?" "Y a-t-il des pics dans ces données de ChIP-seq ?" "Quelle proportion de promoteurs chevauchent des îlots CpG ?" Voilà le genre de questions rencontrées fréquemment en bioinformatique. Nous pouvons y répondre à l'aide de la…
-
Choisir entre R et Python en bioinformatique ? Regards croisés entre collègues enseignants-chercheurs

Gaëlle Lelandais et Pierre Poulain Qui sommes-nous ? Tous les deux passionnés par l’enseignement, les problématiques de big data et d’analyse de données en biologie, nous nous côtoyons professionnellement depuis 15 ans, avec écoute et bienveillance. Si l’étiquette de « bioinformaticien » nous est souvent attribuée, nous sommes pourtant très différents. Je (Gaëlle) travaille sur des problématiques de…
-
Pourquoi et comment déposer un package R sur Bioconductor ?

Ça y est, votre code R un poil brut commence à avoir de la substance et vous envisagez d'en faire un outil à part entière. Comme tout bioinformaticien qui se respecte, vous envisagez donc de packager (ou paqueter en français) proprement cet ensemble de scripts R. Non on ne largue pas une nuée de scripts…
-
Créer des Heatmaps à partir de grosses matrices en R
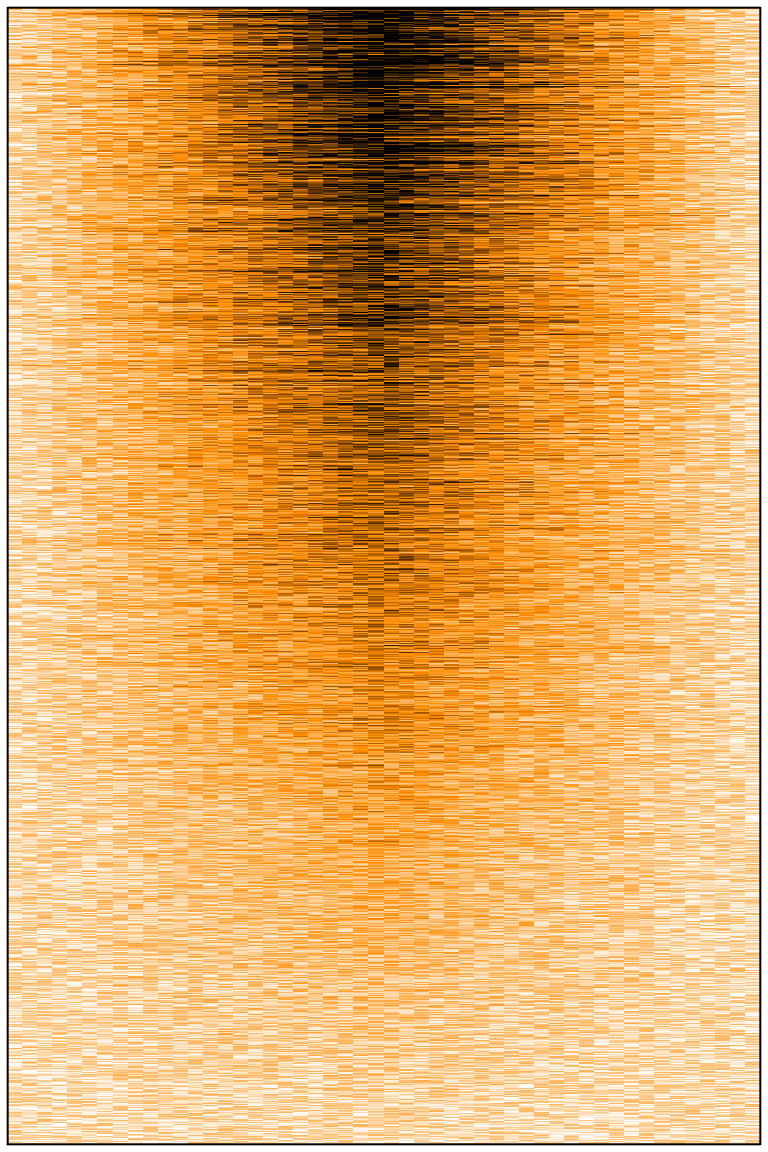
En génomique, et sans doute dans tout un tas d'autres domaines omiques ou big data, nous essayons souvent de tracer des grosses matrices sous forme d'heatmap. Par grosse matrice, j'entends une matrice dont le nombre de lignes et/ou de colonnes est plus grand que le nombre de pixels sur l'écran que vous utilisez. Par exemples,…