

Un jour, un biologiste se pointe chez vous avec d'une part un disque dur externe dans la main, d'autre part l'air soucieux. Il veut que vous analysiez ses données RNA-seq. Le disque, c'est parce qu'il a environ 50Gb de données à vous transmettre ; l'air soucieux, c'est parce qu'elles ont coûté dans les 15'000 euros, et qu'il espère que pour une fois il a pas trop foiré ses manips. Il compte sur vous, vu qu'il n'a aucune idée de ce qu'il a entre les mains, mais il veut des p-valeurs en sortie, et des petites, s'il vous plaît.

Son but peut être multiple :
- démontrer que certains gènes sont transcrits ;
- démontrer que certains gènes sont transcrits dans une condition, mais pas dans l'autre ;
- démontrer un splicing alternatif (ou "épissage") de ces gènes ;
- etc.
Le second est le plus courant. Les "conditions" peuvent représenter la présence/absence d'un facteur de transcription, la présence ou non d'un phénotype/maladie, des environnements de culture différents, des tissus cellulaires différents, etc. Le but est alors de montrer par exemple qu'un facteur de transcription favorise une certaine maladie lorsqu'il est exprimé dans le foie.
Il est aussi censé savoir vous dire sur quelle espèce il travaille, si c'est du "single-end" ou du "paired-end" (voir plus bas), et ce à quoi il s'attend dans les résultats.
Table des matières :
- Introduction
- La préparation
- Le Contrôle de Qualité ("QC")
- L'alignement ("mapping")
- Le comptage
- La visualisation
- Le calcul d'expression différentielle
- L'interprétation
- Conclusion
C'est quoi déjà, un RNA-seq ?
Rappelons la méthode expérimentale [1]: on récolte les ARN de nombreuses cellules à la fois, on les coupe en fragments qu'on amplifie par PCR et qu'on donne à manger à un séquenceur. Celui-ci nous retourne de petits bouts de séquence génomique, des "reads", sous forme de texte avec des A,T,G,C. Vous héritez de dizaines de millions de ces petites phrases.
On part du principe que le nombre de reads est proportionnel à l'abondance des ARN correspondants dans la cellule, le but étant d'estimer cette abondance.
Le RNA-seq est une technique relativement récente et faire partie de ce qu'on appelle "séquençage de seconde génération" (next-generation sequencing) ou "séquençage à haut débit" (high-throughput sequencing), avec le ChIP-seq et ses contemporains.
Remarque : Les ARNm que l'on étudie avec le RNA-seq sont matures - c'est-à-dire poly-adénylés et déjà splicés. C'est un point important, car il se peut qu'un gène produise une grande quantité d'ARN, mais que ceux-ci soient rapidement dégradés avant leur maturation. Or, certains de ces ARN peuvent tout de même être réactifs, par exemple comme facteurs de transcription d'autres gènes. Les ARN immatures, ou "naissants", peuvent être étudiés à l'aide d'autres technologies, comme le NET-seq [2].
Et ces données, elles ressemblent à quoi ?
Les fichiers qu'il vous transmet sont le plus souvent au format FASTQ (texte). Ce pourrait être des SRA (binaire), mais ce format apparaît plutôt en fin d'analyse, au moment de la mise en ligne publique des données. Un FASTQ, ça ressemble à ça :
|
1 2 3 4 5 6 7 8 9 |
@HWI-ST865:166:D0C4KACXX:2:1101:1042:1954 1:Y:0 : CNANAAATNAANNNNGNNNNNNNNNANNNNNAAANNNTNNNNNNNNNTNNTGNNNNTTGTTTNNTTGTGGGTTTCTCTGTCCCCN + ##################################################################################### @HWI-ST865:166:D0C4KACXX:2:1101:1241:1970 1:N:0 : CCAGCGACACTTGCAGCTTAGGGGCAAGAGGCTCCCACAACACCCTGTGCGATCGGAAGAGCGGTTCAGCAGGGATGCCGCGGCC + GFFIGIIIFGEHHIJJJIIGGGHIIBD=BFG?EDECC@FGCHC?BCCBB)53(;;B;?8299?###################### … |
Chaque bloc de 4 lignes, commençant par un "@", représente un "read": la séquence d'un fragment d'ARN de longueur fixe, la qualité (fiabilité) du séquençage de ce morceau, et diverses autres infos illisibles. Vous remarquez par la même occasion que le séquenceur produit des reads courts ("short read sequencing"), d'environ 30 à 150nt.
Remarque : Vu la taille de ces fichiers (des centaines de millions de lignes), un échantillon est souvent divisé en plusieurs fichiers de taille égale.
Ca peut être du "single end", où chaque read est indépendant des autres, ou du "paired-end", où les reads vont par paires, un sur chaque brin, à une distance moyenne fixe l'un de l'autre. Le paired-end est nécessaire pour bien détecter le splicing, et dans un tout autre contexte, les indels. Pour le reste, le single-end fait à peu près aussi bien pour moins cher. Certaines techniques single-end permettent aussi de conserver l'information du brin d'origine (référence ou complémentaire), et donc le sens de transcription.
1. La préparation
- Copier les données à un emplacement qui a beaucoup d'espace disponible et le plus possible de mémoire vive - l'accès à un serveur de stockage et à un cluster de calcul est idéal.
- Se procurer la séquence génomique de l'espèce concernée en format fasta (.fa), par exemple depuis NCBI. Elle sert à différent programmes pour construire des "index" qui servent au mapping (voir ci-dessous).
2. Le Contrôle de Qualité ("QC")

Il faut s'assurer tout d'abord que le séquenceur a bien fait son travail. Les premières paires de bases d'un read sont séquencées avec beaucoup de fiabilité, mais plus on avance dans la séquence, moins c'est précis. Un programme comme FastQC produira divers graphes permettant de savoir s'il faut rétrécir les reads, et de quelle longueur - le "trimming".
Cette étape est très importante puisque des reads de mauvaise qualité s'aligneront difficilement sur le génome, et rendront toute l'analyse inutile : on a déjà vu passer un échantillon de 2% de reads mappés à plus de 60% après trimming - et le doctorant passer des larmes au soulagement.
3. L'alignement ("mapping")
A priori, on ne sait pas de quelle partie du génome provient un read, mais uniquement sa séquence. On va donc "aligner" chaque read sur le génome de l'espèce, c'est-à-dire rechercher dans le génome la position d'une sous-séquence similaire à celle du read. Celle-ci devrait en théorie être unique pour un read suffisamment long (>30nt), et donc on obtient la position d'origine du transcrit d'où il vient.
On imagine le génome comme une grande ligne droite, longue de quelques milliards de caractères, et les reads comme de petits segments venant s'aligner le long de cette ligne, là où la séquence est similaire. Si un transcrit a été séquencé entièrement, tous ses exons seront "recouverts" par des reads, et même plusieurs fois.
Le nombre moyen de reads par position mappée s'appelle "profondeur de séquençage" ("sequencing depth"). C'est pour garantir une profondeur suffisante qu'on doit séquencer des centaines de millions de reads, et s'assurer de leur qualité, car c'est ce qui donne la puissance statistique au moment de déterminer le taux d'expression d'un transcrit.
L'alignement n'est jamais parfait, à cause des erreurs de séquençage, de la phase d'amplification, des régions répétées, des mutations, etc. On trouvera des reads dans des régions non transcrites, mais aussi dans des introns (parfois un ARN n'est pas splicé). Le génome de référence lui-même n'est pas exact. Il est le résultat d'un processus d'assemblage, qui fait également appel au séquençage, et qui est un consensus entre une dizaine d'individus particuliers.
3.1. En partant d'une annotation existante
Supposons qu'on a déjà des fichiers (GTF, BED, etc.) nous indiquant les coordonnées de chaque exon/gène/transcrit. On peut trouver ce genre d'annotation sur Ensembl (BioMart) ou NCBI.
Plusieurs programmes permettent d'aligner des (short) reads : Bowtie, BWA, mais aussi une ribambelle d'autres (souvent commerciaux). Ils se valent, Bowtie et BWA étant les algorithmes les plus rapides et les plus répandus. Nous n'avons pas testé personnellement les programmes payants.
Parmi les options les plus importantes, on veut contrôler le nombre d'alignements reportés (si un read map à plusieurs endroits), et le nombre d'erreurs dans la séquence ("mismatches") permises. En effet, le séquenceur possède un taux d'erreur d'environ 1%, donc on s'attend à au moins 1 erreur sur un read de 100nt. De plus, bien que des séquences de 100nt soient uniques en théorie - si la séquence du génome était aléatoire, - il existe de nombreuses région répétées ou très similaires.
3.2. En reconstruisant l'annotation
Si on ne fait pas confiance aux annotations disponibles, ou qu'on cherche de nouveaux exons/isoformes, on peut reconstruire complètement ou partiellement la structure du génome grâce à nos reads, au moment de l'alignement. Des programmes comme TopHat ou Cufflinks savent deviner la limite des exons et reconstruire les isoformes possibles à partir des données.
Si on reconstruit ainsi de novo l'entier du génome, cette approche est en revanche très lente et requiert une grande quantité de mémoire. Un bon compromis est de leur fournir une annotation (sous forme de fichier GTF) qu'ils se chargeront de compléter.
3.3. Le résultat
Dans tous les cas, le résultat de cette étape est un fichier BAM (version binaire) ou SAM (version texte). Le SAM est rarement utile et prend vraiment trop de place, c'est pourquoi on utilise en principe seulement le BAM, qu'on manipule avec samtools. Dans les deux cas, le contenu est identique est ressemble à ça :
|
1 2 3 |
HWI-ST865:166:D0C4KACXX:4:1310:3832:30957 16 chr20q 1348 255 159M * 0 0 GCATGAAAACATCCTAGGAGTCCTGTGCAGGGAATTTTATTTTGGACTTCTTCACATTCCTGTCTCTGTCTCAAGTCTCCACCTGTTCTCTTTGTCTGGTTTTCCTGCTACTGCCTTAGGTCCCCGACTTGCCACACTTAGGCTTGTGGGACCTCCTCA HHHJIJJJJGIJIJIIJJJJIJJJJJJJJJIJIJJJJJJIGIJJJJJJJJJJJJJIJJIJJJHHHHHHGFFFFFFDEEEEDEDDDCCDDCCCB?DA?<ACC@DDEDCEEDFFFHHHHDJIGJJJIHGJIJJIIIIHJJJJJJJJJJJJIGCJIIJJGHH XA:i : MD:Z:159 NM:i:0 NH:i:2 HWI-ST865:166:D0C4KACXX:4:1216:20673:42398 16 chr20q 1900 255 151M * 0 0 GCCTTCCCAGACTTTCCCTTTTCTAGGAGCTCCCCTCCTGTTCATGTCTAGCTATATATAAACTCTAACAGAGCCCACTATCCTGTGTCTTTCCAAAAATAGTGAGGGAATGATTAATTGGAAACCATAAGAAGTGATATGCATGTAGATG HHHEGGGGEGIGGIHIHGHGIHIIGGIHIGEAHGIJI7D?DDGDHGFFHJIGHAGGHGDEEC=@GHAAE>CCCCEC@EECEACCC=EGEC7:BHB<CHD?<?G>GEDBFHCCIFHDD<CIHFEEIIIGHIHGHCGGGGGGIIHBHIIGHDH XA:i:0 MD:Z:151 NM:i:0 NH:i:1 … |
Ici chaque ligne représente un alignement : l'identifiant du read, le nom de la séquence de référence, la position de l'alignement sur la référence, la séquence du read, la qualité de l'alignement, et divers indicateurs qualitatifs ("flags"). Pour plus de détails, voir la spécification du format SAM.
Remarque : la qualité de l'alignement n'a rien à voir avec la qualité du read précédemment décrite. Ici elle décrit combien on est sûr que le read provient de cette position du génome.
3.4. Cufflinks & autres
Le nom de Cufflinks apparaîtra souvent dans cet article. En effet, il fait à peu près tout, avec l'inconvénient d'être réputé pour
- avoir un output compliqué, voire décourageant, en tout cas à première vue ;
- produire des bugs qui vous tuent un job après 3 jours sur le cluster (d'après certains collègues);
- ne pas être personnalisable puisque ce n'est pas votre code.
A vous de voir, on vous recommande toutefois de le tester dans sa dernière version. A noter que d'autres alternatives sont publiées à l'heure de cet article, et que d'autres le seront encore. On peut citer par exemple RSEM.
4. Le comptage

Sous l'hypothèse que le nombre de reads venant d'un certain gène est proportionnel à l'abondance de son ARN dans la cellule, on veut compter les reads venant de chaque gène, transcrit ou exon du génome.
4.1. La préparation d'une table
Partant de nos fichiers BAM, une solution est d'utiliser samtools dans un script maison. En ligne de commande, "samtools mpileup" fournit par exemple un format exploitable ; le wrapper Python de samtools, pysam, permet plus de flexibilité et d'efficacité si on code dans ce langage. Il existe d'autres wrappers pour différents langages (R, Ruby, C++, Java, Perl, Haskell, Lisp).
En fait, si on fait cette partie soi-même (ce qui est justifiable), on se dépatouille comme on peut, pour autant qu'on produise un fichier texte lisible - et déjà utile au biologiste, - qui ressemble déjà à une table, avec un header explicite, du genre qu'on peut charger dans R facilement. Du type :
|
1 2 3 4 5 6 7 8 9 10 |
GeneID,counts1,counts2,Start,End,GeneName,Strand,Chromosome ENSMUSG00000028180,134.0,135.0,157197123,157211354,Zranb2,1,chr3 ENSMUSG00000002010,426.0,526.0,71024301,71032236,Idh3g,-1,chrX ENSMUSG00000002017,301.0,111.0,75936425,75951286,Fam98a,-1,chr17 ENSMUSG00000028184,565.0,478.0,148478549,148652280,Lphn2,-1,chr3 ENSMUSG00000002015,497.0,923.0,70931516,70961514,Bcap31,-1,chrX ENSMUSG00000028186,12465.0,11435.0,146260040,146295269,Uox,1,chr3 ENSMUSG00000028189,69.0,64.0,146113433,146128813,Ctbs,1,chr3 ENSMUSG00000040147,792.0,891.0,16286407,16394492,Maob,-1,chrX ENSMUSG00000053219,22.0,22.0,21893326,21903720,Raet1c,1,chr10 |
Sinon, une librairie Python, HTseq, permet de faire la même chose si on arrive à comprendre sa documentation. Cufflinks, à nouveau, sait le faire tout seul aussi, que ce soit à partir d'une annotation complète ou avec ce qu'il construit lui-même.
4.2. Le problème des jonctions
En alignant sur le génome ou l'exome, les reads qui proviennent d'une jonction de splicing (à cheval sur deux exons réunis après splicing de l'ARN) ne retrouveront plus leur origine, puisque deux moitiés de la séquence du read sont séparées par un intron dans la référence. C'est triste, et puis avec ça on perd une partie des données. Pour y remédier, on peut
- réaligner les reads n'ayant pas pu être alignés sur le génome, sur le transcriptome, et additionner ces reads aux exons correspondants ;
- utiliser un programme spécialisé, comme SOAPsplice ou TopHat, avec les reads non alignés sur le génome, pour détecter les jonctions ;
- lancer un programme comme TopHat ou Cufflinks avec tous les reads (très long).
Les deux derniers fonctionneront infiniment mieux avec du paired-end, puisqu'un écart exagéré entre deux reads d'une paire sera la preuve qu'une partie d'un transcrit a été coupée, comme c'est le cas lors du splicing.
4.3. Le problème des transcrits alternatifs
Quand on cherche à quantifier l'expression des différents transcrits alternatifs (isoformes) d'un gène, on ne peut pas simplement compter combien de reads tombent entre les coordonnées génomiques de chacun, qu'on peut trouver sur Ensembl. En effet plusieurs d'entre eux partagent les mêmes exons, et si un read mappe sur un exon commun, il est impossible de savoir de quel transcrit il provient. Pour plus de détails, voir cet autre article de notre blog.
On peut toutefois essayer de distribuer les reads de manière optimale entre les isoformes, ou d'un autre point de vue, chercher la distribution la plus probable. Plus concrètement, des solutions possibles sont
- Les moindres carrés (avec contrainte de positivité, "non-negative least-squares") pour résoudre de manière optimale le système linéaire surdéterminé
, où
est le vecteur des comptes par transcrit,
celui des comptes par exon, et
la matrice de correspondance indiquant (avec des 1 et des 0) quel exon se retrouve dans quel transcrit.
- La méthode du maximum de vraisemblance appliquée à la somme des probabilités de chaque read d'appartenir à tel ou tel transcrit. C'est la méthode implémentée dans Cufflinks, par exemple.
- Un algorithme EM, qui fait l'objet de plusieurs publications récentes (RSEM, iReckon).
Ces méthodes donnent des résultats assez différents, la première, plus équitable, appliquant plutôt "le bénéfice du doute", tandis que la seconde tend à privilégier un transcrit particulier. Nous n'avons pas encore pu tester la troisième. On ajoute parfois un LASSO pour "corriger" certains défauts de la méthode.
Il est très difficile de savoir laquelle est la plus réaliste, n'ayant pas beaucoup de cas (pas du tout?) de gènes dont on a mesuré par un autre moyen l'expression relative des isoformes (qui plus est pour votre type de cellule, dans vos conditions, etc.). Les résultats sont donc assez hypothétiques, et la solution à ce genre de questions viendra probablement d'un autre type d'expérience que le RNA-seq. On peut cependant utiliser la qRT-PCR pour estimer plus précisément l'abondance d'un petit nombre de transcrits, ou faire des simulations. Le sujet est encore très ouvert.
4.4. Les duplicats PCR
Durant l'étape d'amplification des fragments d'ARN (PCR) qui précède le séquençage, certains fragments peuvent être sur-amplifiés par erreur. Le résultat est un nombre de copies anormal de reads identiques, qui peut biaiser les résultats. Heureusement, de tels "duplicats PCR" sont assez facilement repérables après visualisation (section suivante), puisqu'on observe des colonnes disproportionnées de plusieurs centaines de reads identiques superposés. C'est beaucoup plus difficile à détecter et en amont et sur l'ensemble des gènes, ce qui pose évidemment un problème statistique au moment de la comparaison entre échantillons, si un fragment est sur-amplifié dans l'un et pas dans l'autre. Il n'existe par contre pas encore à notre connaissance de bon outil pour filtrer ces duplicats de manière systématique.
5. La visualisation
5.1. Genome browsers
Pour avoir déjà un aperçu de ses données, le mieux est de construire pour chaque échantillon ce qu'on appelle un "profil" ou une "densité" du signal, c'est-à-dire une sorte de "bar plot" qui à chaque position du génome indique le nombre de reads mappés à cette position. Cela se présente sous la forme d'un fichier bien formaté qu'un programme de visualisation ("genome browser") comme UCSC (en ligne) ou IGV (local) saura interpréter pour produire un graphique.

des CpG-islands, des éléments répétés, les gènes Refseq et Ensembl,
le GC content et des pics de ChIP-seq.
C'est extrêmement utile pour plusieurs raisons :
- On voit les "montagnes" de reads qui localisent les exons et autres transcrits.
- On peut comparer visuellement les niveaux d'expression de plusieurs échantillons, et des gènes entre eux.
- On peut ajouter des profils d'autres types de signaux, comme du ChIP-seq, de la méthylation, de la conservation, des motifs, etc. pour expliquer notre signal. C'est une approche très moderne et qui est souvent nécessaire à justifier une découverte : si un gène est exprimé dans une condition et pas dans une autre, on s'attend à observer de la Polymérase II, un pic de H3K4 méthylé au début, un pic de machin au milieu, un pic de truc à la fin.
Pour obtenir ces densités, il existe une multitude de programmes que chaque labo de bioinfo a recodé pour lui… La fonction "genomeCoverageBed" de bedtools peut apparemment le faire, sinon téléchargez ou codez vous-même un n-ème "bam2wig", sachant que ça prend du BAM en entrée (à nouveau, on peut utiliser samtools ou son wrapper pysam) et que ça doit ressortir un bed/bigBed/bedGraph/wig/bigWig.
Pour les visualiseurs en ligne, sachez tout d'abord qu'il y a moyen de voir les BAM directement, et ensuite que les formats préférés sont ceux où il n'y a pas besoin de télécharger tout le fichier : on upload seulement un "index", et quand on fait une requête pour une nouvelle région, ça vient télécharger depuis votre ordi seulement la partie qu'il faut. C'est le cas des bigBed/bigWig, formats d'ailleurs inventés pour le browser UCSC qui fournit les outils de conversion (p.ex. bedGraphToBigWig), et des BAM. Ce sera alors énormément plus rapide.
5.2. MA-plot
Un type de graphe est aussi fréquemment utilisé pour comparer deux échantillons, une fois les comptes obtenus : le MA-plot. La lettre M représente les log-ratios, et la lettre A est pour "Average" (la moyenne). Comme son nom l'indique, le MA-plot est un plot de la moyenne (en x) contre le ratio (en y) de deux valeurs : l'expression d'un gène dans deux conditions, chaque gène étant un point du graphe.
C'est-à-dire : si 




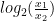

(Ici des quasi réplicats). Source : HTSstation
Il s'interprète comme suit : plus un point est à droite du graphe, plus le gène est globalement fortement exprimé, et inversement. Plus un point est en haut du graphe, plus il est surexprimé dans la première condition par rapport à la seconde, et inversement. Les gènes "intéressants" se trouvent donc plutôt vers la droite (ratios plus convaincants si les comptes son élevés) et le plus possible en haut ou en bas du graphe, visiblement séparés de la masse des autres points.
Deux réplicats techniques montreront par exemple très peu de variation autour de l'axe horizontal, alors que deux échantillons assez différents donneront au graphe une forme de gros poisson. On peut vérifier aussi que la médiane est bien à zéro, sans quoi on observe un biais : les échantillons seraient mal normalisés (voir plus loin).
C'est une approche purement visuelle qui ne démontre rien, mais si un gène qu'on suppose jouer un rôle important se démarque facilement sur ce graphe, on sait déjà qu'on a réussi, et on peut ajouter une très jolie figure à la publication, ce qui n'est pas non plus négligeable.
La librairie R limma sait le faire, mais on peut facilement produire son propre script, tel ce package Python, qui a l'avantage d'être interactif : si on clique sur un point, le nom du gène associé s'affiche.
6. Le calcul d'expression différentielle
Là, ça se complique. Pour un gène donné, on veut savoir s'il est significativement plus exprimé dans une condition que dans l'autre.
Par exemple, si j'ai 100 reads dans la condition 1, et 500 dans la condition 2, on dirait que oui (mais en fait, on n'en sait rien). Et s'il y en a 10 et 50 ? 1 et 5 ? la proportion est la même, et pourtant ça pourrait être dû au hasard. Et s'il y en a 100 et 150 ? Là, on a besoin des mathématiques pour se convaincre.
6.1. La normalisation
Quand on veut comparer des échantillons, il faut d'abord les rendre comparables. En effet, des biais comme le nombre total de reads alignés pour un échantillon, des artéfacts PCR, etc. peuvent fausser les résultats. Par exemple, imaginons qu'il y a 10 fois plus de reads dans une condition que dans l'autre, alors la quasi totalité des gènes sera déclarée significativement enrichie.
Pour y remédier, on a pensé à diviser chaque échantillon par le nombre total de ses reads. Malheureusement, comme en RNA-seq de petites régions ont tendance à accumuler une grande partie des reads, cela crée plus de biais que ça n'en corrige. Imaginez deux échantillons où l'un a deux fois plus de reads que l'autre, mais où la moitié est contenue dans un seul gène. Alors en divisant par deux, on divise tous les gènes par deux, et tous deviennent significativement moins exprimés…
On peut aussi considérer de diviser les comptes dans chaque gène par la taille de ce gène, pour obtenir une "densité" de reads. C'est alors un autre point de vue : cette transformation est nécessaire pour comparer des gènes d'un même échantillon entre eux, mais pas pour comparer différents échantillons. Notons cependant que certaines publications [3] montrent un biais dans la détection de gènes différentiellement exprimés en faveur des gènes les plus longs. Malgré une perte de puissance au niveau statistique, il pourrait être préférable de diviser les comptes par la longueur des transcrits (?).
En alliant les deux, on obtient des RPKM, pour Reads Per Kilobase and per Million (of reads). La formule est donc 
Une meilleure solution est par exemple présentée dans l'article lié à DESeq [4] dont nous parlerons plus loin. En gros, on construit une nouvelle "référence" en prenant la moyenne (géométrique, plus robuste) des échantillons et en les normalisant tous par rapport à cette référence.
Remarque : en fait, le RNA-seq ne permet pas de différencier une amplification naturelle de l'expression de tous les gènes d'un individu, d'un artefact technique d'amplification. Dans le premier cas il ne faudrait pas normaliser, dans le second, si. On part toujours de l'hypothèse que le niveau d'expression de la plupart des gènes ne change pas entre deux échantillons - la plupart du temps.
6.2. Les stats

pour différentes valeurs de lambda.
(source : Wikipedia)
|
1 |
Maths ON |
Chaque read a un chance 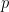
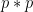
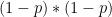
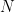
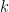


Heureusement, on peut approximer une binomiale avec une loi dite de Poisson. Une formule nous donne alors la probabilité d'observer un certain nombre de reads (connu), si on connaît un paramètre 
Remarque : en réalité, on observe que la loi de Poisson est un peu trop restrictive, et on utilise plutôt une loi "négative binomiale", façon d'ajouter un terme de variance supplémentaire. Un changement qui amène des complications puisqu'il faut estimer cette variance à son tour…
[|
1 |
Maths OFF |
Rassurez-vous, un programme le fera aimablement à votre place, donc au pire vous pouvez joyeusement oublier tout ça et appuyer sur le bouton rouge.
Actuellement, les programmes le mieux à même de faire ces calculs sont des librairies R du nom de DESeq et EdgeR. Tous deux possèdent un manuel très clair, un article lisible et une référence complète. Le programme, donnée votre table obtenue précédemment, vous retourne un nouveau fichier tabulé avec une belle colonne de p-valeurs ajustées, dont vous pouvez extraire une liste des gènes les plus significativement sur-/sous-exprimés dans une certaine condition. Il se charge aussi de la normalisation si nécessaire de votre table avant de faire quoi que ce soit.
Cufflinks prétend aussi faire tout cela très bien dans sa nouvelle version, ce que nous n'avons pas testé. Le modèle mathématique est globalement le même dans tous les cas.
7. L'interprétation
A présent, il faut voir si les gènes qui ont été trouvés significatifs ont quelque chose à voir avec l'expérience. Une liste de 100 gènes, en soi, n'est pas très utile. Souvent le biologiste saura indiquer quels gènes il s'attend à trouver dans la liste, et on espère qu'ils sont dans les premiers. Si au contraire il ne sait pas ce qu'il cherche, il faut être conscient que la démarche est exploratoire, et des arguments Bayesiens [5] montreront que les résultats n'ont en aucun cas valeur de preuve.
Enfin, si les p-valeurs ajustées sont trop grandes, (c'est nul et) on peut toujours filtrer par p-valeur simple, voire par "fold change" (ratio) pour faire croire au biologiste que non, tout espoir n'est pas perdu. L'effet est particulièrement observé en l'absence de réplicats.
Si c'est à peut près tout ce qu'on peut tirer du RNA-seq lui-même, l'analyse peut faire intervenir d'autres types de données, qui rendent le jeu plus complexe et probablement plus intéressant. Des contraintes particulières peuvent aussi vous obliger à changer vos paramètres de mapping, vos limites de p-valeurs, etc.
8. Conclusion
Le RNA-seq est une méthode relativement récente, et par conséquent les outils disponibles non seulement ne sont pas aussi au point que pour les microarrays, mais qu'il évoluent rapidement ou sont régulièrement remplacés par d'autres - en particulier ceux qui proposent un pipeline complet. Cependant c'est une technique prometteuse, qui se présente comme le successeur des microarrays en livrant plus d'informations structurelles tout en ayant une sensibilité comparable ou supérieure, déjà avec les outils d'analyse existants. Son principal défaut étant son coût, le fait que celui-ci baisse très rapidement la rend de plus en plus populaire, et donc rend de plus en plus nécessaire la mise au point de pipelines efficaces.
Et voilà.
Références de l'article :
[2] L. S. Churchman, "Nascent transcript sequencing visualizes transcription at nucleotide resolution", Nature 2011 [3] A. Oshlack, "Transcript length bias in RNA-seq data confounds systems biology", Biology Direct 2009 [4] S.Anders, "Differential expression analysis for sequence count data", Genome Biology 2010 [5] J.P.A. Ioannidis, "Why Most Published Research Findings Are False", PLOS Medicine 2005
[1] A. Mortazavi, "Mapping and quantifying mammalian transcriptomes by RNA-Seq", Nature Methods 2008
Les images ne possédant pas de sources sont de l'auteur de l'article.
Merci à Clem_, Hautbit, nahoy, ZaZo0o et Yoann M. pour leur relecture.
Laisser un commentaire