

Qui sommes nous ?

Une communauté d'étudiants et de professionnels

Une base d'articles libres pour tous niveaux

Des salons de discussion actifs au quotidien
-
L'alignement multiple en étoile ⭐
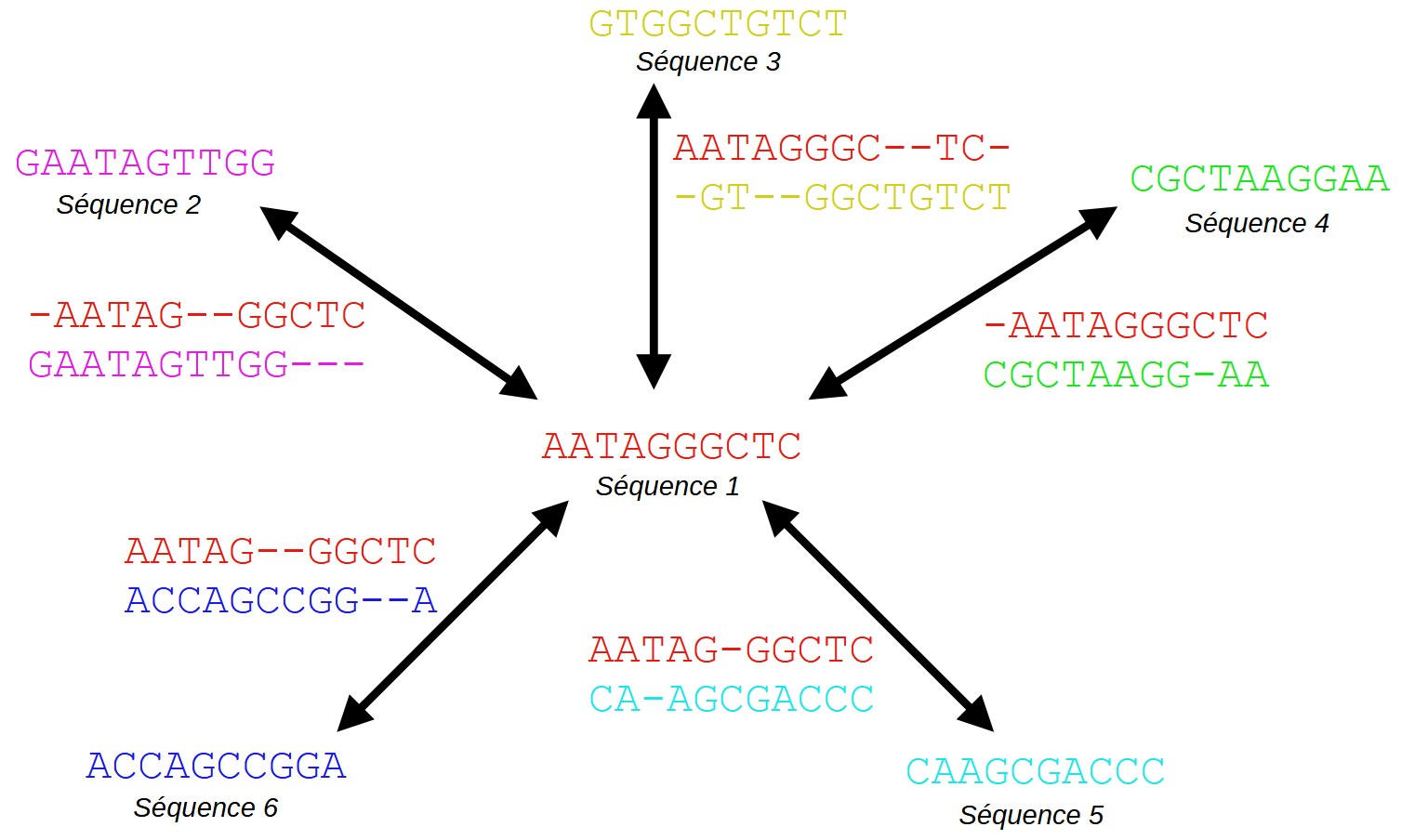 Lire la suite : L'alignement multiple en étoile ⭐
Lire la suite : L'alignement multiple en étoile ⭐Introduction Dans le domaine de la bioinformatique l'alignement de séquences est une opération courante pour comparer des séquences biologiques (ADN, ARN, protéines). Nous avons vu dans un précédent article le principe de l'algorithme de Needleman-Wunsch qui produit un alignement global optimal de deux séquences. Au-delà de l'alignement par paire, l'alignement multiple de séquences (Multiple Sequence […]
-
Bonne rentrée 2025 à tous 🎓
 Lire la suite : Bonne rentrée 2025 à tous 🎓
Lire la suite : Bonne rentrée 2025 à tous 🎓À tous les p'tits veinards qui ont profité de vacances cet été 🌴— et aux autres aussi — on vous souhaite une belle rentrée 🎓. C’est cette période de l’année où l’on prend un nouvel élan : on planifie, on se projette, on fixe de nouveaux objectifs. Peut-être un nouveau poste pour certains, une formation pour […]
-
Licence Bio-Informatique, Statistique et Modélisation (BISM) à Lyon 1
 Lire la suite : Licence Bio-Informatique, Statistique et Modélisation (BISM) à Lyon 1
Lire la suite : Licence Bio-Informatique, Statistique et Modélisation (BISM) à Lyon 1Cet article est une mise à jour de https://bioinfo-fr.net/presentation-de-la-licence-miv-a-lyon-1. Cette formation pluridisciplinaire existe depuis 2010 (ex. Modélisation et Informatique du Vivant), et bénéficie de la recherche historique lyonnaise en analyses de données biologiques et notamment en biométrie et statistiques. Les aspects liés à la bioinformatique ont été renforcés à partir de 2018, en lien avec […]
-
Alignement de séquences : bien comprendre l'incontournable algorithme de Needleman-Wunsch
 Lire la suite : Alignement de séquences : bien comprendre l'incontournable algorithme de Needleman-Wunsch
Lire la suite : Alignement de séquences : bien comprendre l'incontournable algorithme de Needleman-WunschIntroduction L'analyse des séquences biologiques (ADN, ARN ou protéines) est un sujet central de la biologie moléculaire et de la bioinformatique. Dans ce cadre, chercher à savoir à quel point deux séquences sont similaires ou dissimilaires est une des questions qu'on se pose le plus souvent (par exemple dans le but d'analyser des relations phylogénétiques). […]
-
Des molécules et des données !
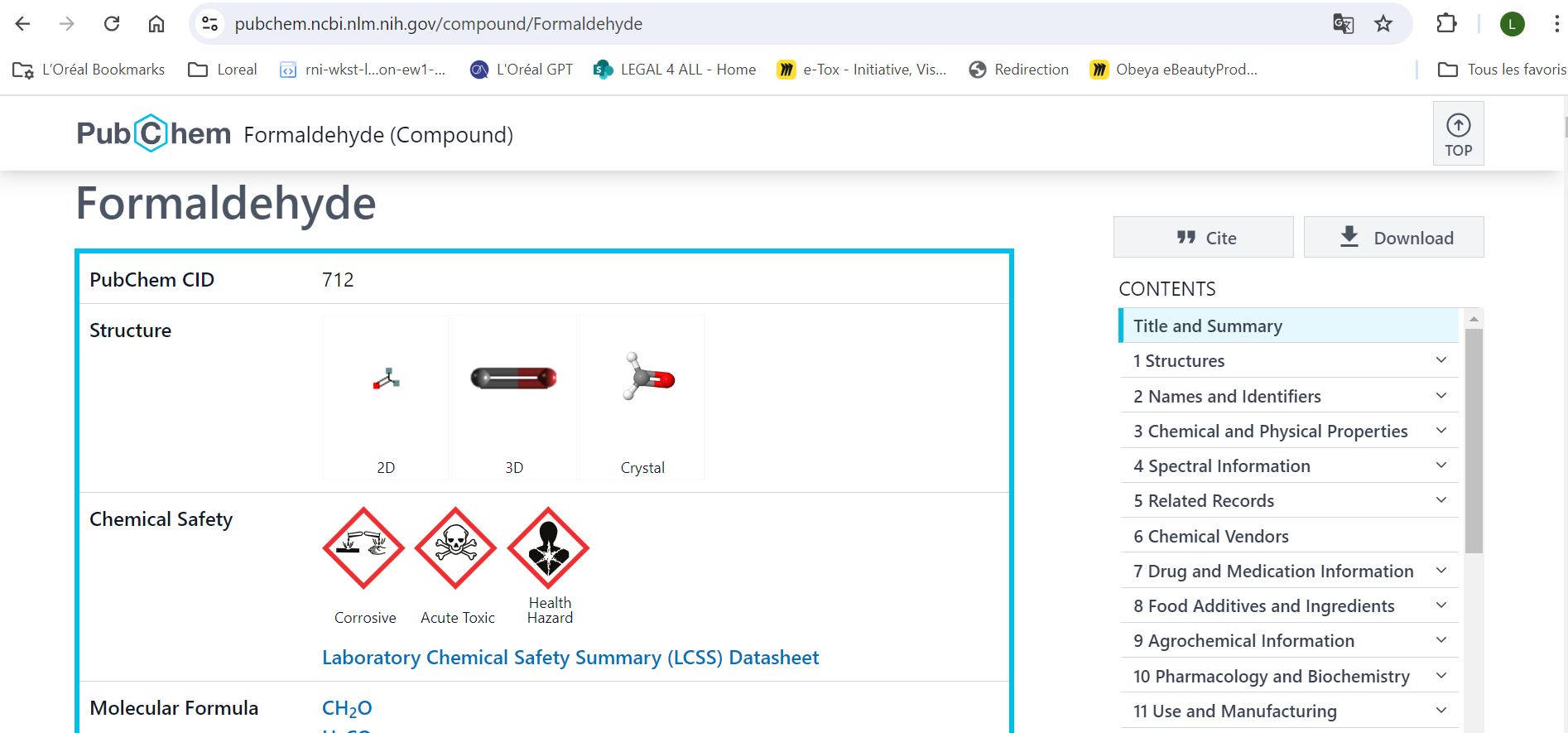 Lire la suite : Des molécules et des données !
Lire la suite : Des molécules et des données !Vu que nous sommes sur un blog de bio-informatique, je vous propose aujourd'hui de parler chimie ! Enfin plutôt, chémoinformatique* et quelques notions de la façon dont une molécule est stockée en ligne en fonction des réglementations chimiques et cosmétiques. Je ne suis pas un expert de ce domaine, mais alors vraiment pas du tout. Par […]
-
Software Heritage : l'archive universelle des codes sources logiciels
 Lire la suite : Software Heritage : l'archive universelle des codes sources logiciels
Lire la suite : Software Heritage : l'archive universelle des codes sources logicielsDans cet article, je vous propose de découvrir Software Heritage, l’archive universelle dédiée à la collecte, la préservation et le partage des codes sources logiciels. En 2021, je suis devenu ambassadeur pour Software Heritage, c’est-à-dire représentant de la communauté bioinformatique, dans l’infrastructure Software Heritage. Pourquoi archiver son code source ? Aujourd’hui, les scientifiques utilisent très souvent des […]
-
Conda et le piège de la licence Anaconda
 Lire la suite : Conda et le piège de la licence Anaconda
Lire la suite : Conda et le piège de la licence AnacondaConda est apparu en 2012 et a été distribué par Continuum Analytics, connue sous le nom d’Anaconda Inc depuis 2017. Conda est distribué sous la licence MIT, l’une des licences open-source les plus permissives, permettant aux utilisateurs d’adopter, d’adapter et de partager le logiciel presque sans restrictions. Conda a été conçu pour gérer les environnements […]
-
ABSD : base de données d'anticorps non redondants et standardisés
 Lire la suite : ABSD : base de données d'anticorps non redondants et standardisés
Lire la suite : ABSD : base de données d'anticorps non redondants et standardisésLes anticorps (immunoglobulines) jouent un rôle crucial dans la réponse immunitaire contre les menaces extérieures, telles les infections virales. Une immunoglobuline est composée de deux molécules en interaction appelées chaîne légère et chaîne lourde : la combinaison d’une chaîne légère et d’une chaîne lourde donne une immunoglobuline (voir Figure 1). Bien que le nombre théorique d'immunoglobulines […]
Articles les plus lus ces 7 derniers jours



Les salons de discussion :


La boutique

Quelques liens
Étiquettes
ADN analyse base de données bioinformatique code concours conférence débutant Découverte edito emploi formation Génomique Interview JeBiF JOBIM master modélisation outil programmation Python R Recherche rentrée script SFBI statistiques séquençage tutoriel vacances visualisation