

Dans la grande famille du séquençage, on distingue le DNAseq et le RNAseq. Le premier capture la séquence d'ADN contenue dans un organisme, un tissu, une tumeur. C'est un peu comme décompiler le code source d'un être vivant pour comprendre comment il fonctionne. Le postulat de l'ADN est qu'il est sensé être le même dans toutes les cellules d'un organisme (à part de rares exceptions). Le RNAseq, quant à lui, permet de capturer le produit de l'expression des gènes à un moment donné dans un tissu spécifique. En effet, les gènes ne s'expriment pas de la même manière d'un tissu à l'autre et également au cours du temps. L'étude du transcriptome permet de savoir exactement quels gènes sont exprimés, s'ils sont beaucoup ou peu exprimés, et s'ils sont bien exprimés (si l'ARN est bien fait, de telle sorte qu'il sera fonctionnel).
Le premier point commun entre ces deux techniques est sans aucun doute le point de départ. Pour faire du séquençage, il faut des échantillons (et avoir bien pensé son expérience, évidemment). On prélève des bouts de tissus directement sur l'organisme (mort ou vivant) et on se lance dans l'extraction de la molécule à séquencer (ARN ou ADN). Selon le type de tissu que l'on va prélever, on va potentiellement extraire un mélange de cellules de différents types. Si l'on prélève du sang par exemple, ce qui est majoritairement le cas des études sur les humains, on va obtenir au moins 5 types de globules blancs différents, plus les globules rouges (mais ceux-ci n'ont pas d'ADN).
Le résultat du séquençage de ces cellules donnera une vue globale de la séquence d'ADN de l'individu où l'on va observer les mutations trouvées dans tous ces types cellulaires. Maintenant, si l'on veut étudier des tumeurs circulantes (cellules qui se déplacent dans le sang),on ne pourra pas savoir quelles cellules sont mutées et lesquelles sont normales, et donc on ne pourra pas interpréter les résultats.
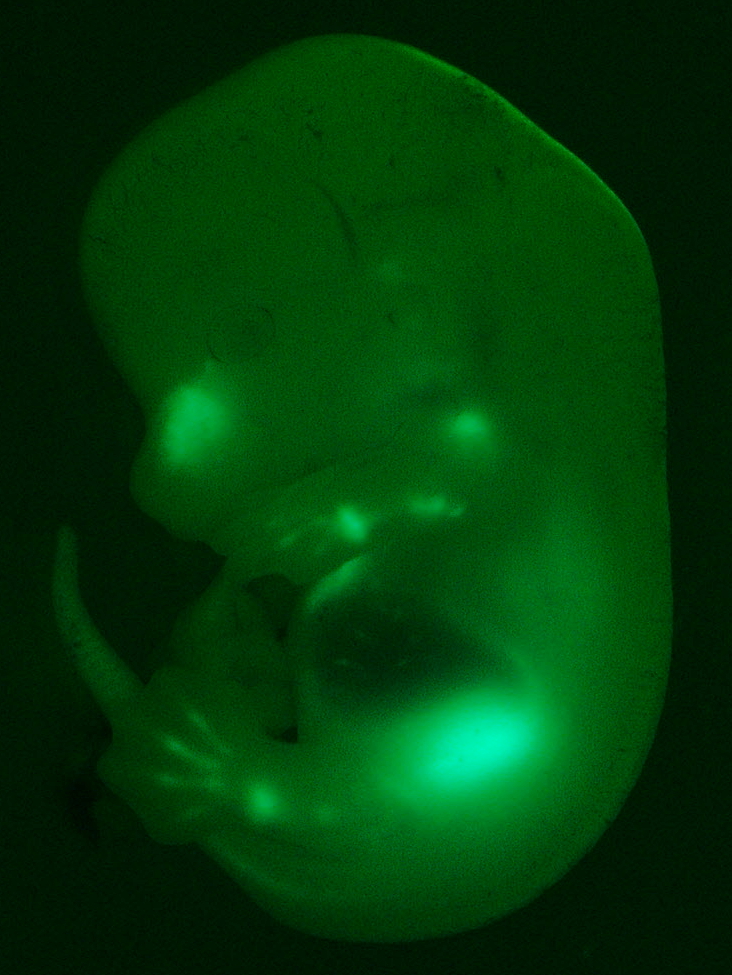
En matière de RNAseq, c'est encore pire, si on peut dire. Chaque type cellulaire se distingue par un profil d'expression de gènes bien spécifiques (bien que l'ADN soit le même). Si l'on mélange plusieurs types cellulaires, on va obtenir un mélange d'ARN dont on ne saura pas distinguer l'origine. On observera l'expression de gènes mais on ne saura pas dire s'ils sont exprimés dans un type de cellule en particulier, ou s'ils sont exprimés un peu chez tout le monde… Selon le but de votre analyse cela peut devenir extrêmement problématique. Quand on a la chance de travailler sur des organismes modèles de laboratoire (bactéries, drosophiles, souris, rats, poissons, etc…), on peut générer des organismes transgéniques chez lesquels on a introduit un gène marqué qui produit une molécule fluorescente (cf. l'embryon de souris vert à droite). On choisi le gène à marquer en fonction de la spécificité de son expression. Il faut que ce gène ne s'exprime que dans le tissu ou le type cellulaire qui nous intéresse. Ensuite, on peut extraire les cellules fluo par cytométrie en flux et hop, on ne séquence que les cellules qui nous intéressent. Mais quand on travaille sur les humains, comment fait-on ?
L'autre paramètre à prendre en compte quand on veut faire du séquençage est la quantité de matériel nécessaire. Pour un RNAseq par exemple il faut environ 200ng d'ARN pour un réplicat, soit pas moins de 40 000 cellules (si je ne me trompe pas). Vous vous imaginez que, dans ces conditions, on ne peut pas séquencer des ovules ou des stades très précoces d'embryons…
En résumé, le DNAseq et le RNAseq, pour la plupart des études, c'est déjà très bien. Mais lorsque l'on veut étudier des phénomènes extrêmement précis ou que l'on est limité niveau quantité de matériel, les techniques traditionnelles révèlent leurs limites. Mais comme le disent les Shadoks, s'il n'y a pas de solution c'est qu'il n'y a pas de problème ! Et la solution ici tient en trois mots : Single Cell Sequencing.
Sur le papier, le principe est simple. On isole des cellules individuellement, on extrait l'ADN ou l'ARN, on amplifie une première fois l'ADN ou l'ARN pour obtenir assez de l'ADNc, et hop, on passe aux étapes classiques de la préparation de séquençage : préparation des librairies, deuxième étape d'amplification et séquençage en lui même.
Dans la pratique, ce n'est malheureusement pas aussi simple. La grosse difficulté est d'extraire les cellules et de les isoler, et ça, ce n'est pas une mince affaire. Plusieurs techniques existent, mais chacune a ses inconvénients (cf. tableau), à vous de choisir celle qui vous convient en fonction de vos moyens. Pour l'amplification, il doit maintenant exister des kits avec des produits bien dosés qui feront le travail pour vous.
| Méthode | Débit | Coût | Manuel/Automatique |
|---|---|---|---|
| Micromanipulation | Lent | Faible | Principalement manuel |
| Fluorescence-activated cell sorting (FACS) | High-throughput | Elevé | Automatique |
| Laser-capture microdissection | Lent | Elevé | Manuel |
| Microfluidics | High-throughput | Elevé | Automatique |
L'analyse de données Single-cell ne s'appréhende pas tout à fait comme une analyse classique. On va certes utiliser la batterie de logiciels habituelle, mais il faut plus de points de contrôle. En général, en parallèle du single-cell, on procède à un RNAseq (ou DNAseq) total du même tissu dont ont été extraites nos cellules afin d'avoir une référence. Ce RNAseq (ou DNAseq) total servira de base pour contrôler la qualité de vos single-cell et aussi pour confirmer ou infirmer l'expression de certains gènes. Dans le meilleurs des cas, vous aurez multipléxé vos séquençages, autrement dit, lors de la préparation des librairies, les ADNc ont été marqués avec un code barre propre à chaque cellule, puis les ADNc sont mélangés et séquencés ensemble. Le multiplexage permet de séquencer plus de choses et à moindre coût vu qu'on remplit au mieux le séquenceur. Si vous êtes dans ce cas, vous pourrez "pooler" les reads des single-cell ensemble pour avoir un simili RNAseq total et vous en servir pour faire une estimation du bruit obtenu. En effet, vu que l'on part avec une quantité réduite d'ARN (ou ADN), deux étapes d'amplification sont nécessaires (au lieu d'une seule normalement). L'amplification peut créer des biais dans vos données et il est important d'en avoir conscience, surtout quand on fait du single-cell. D'ailleurs, à cause de ces bruits générés par une grosse amplification, il va être difficile d'analyser de manière très fine l'expression de gènes faiblement exprimés.
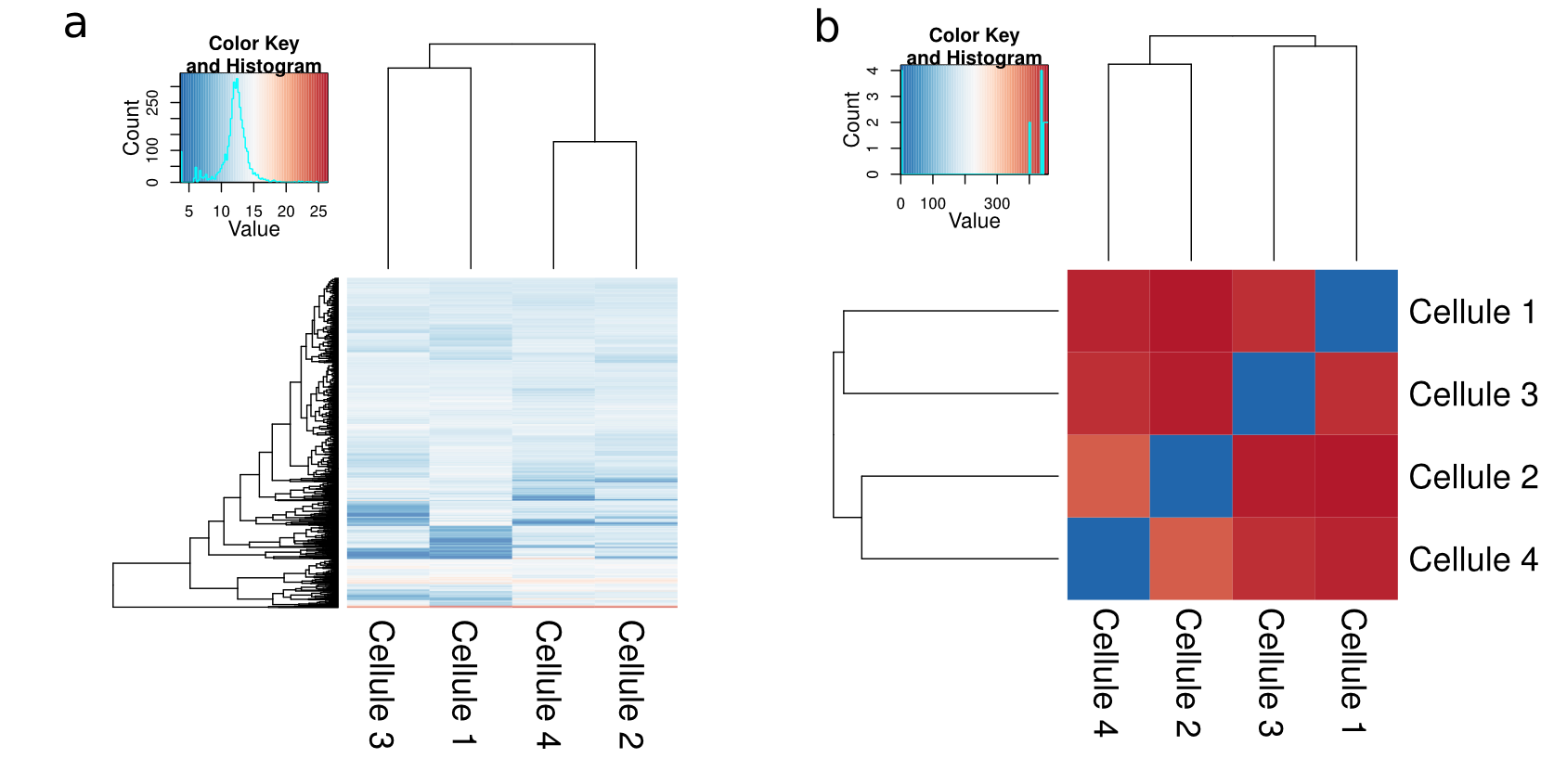
Le second souci du single-cell, c'est la variabilité de l'expression entre les cellules, même de type identique. De prime abord, on pourrait s'attendre à avoir des profils d'expression assez similaires d'une cellule à une autre, surtout quand il s'agit du même type. Or, il faut savoir que ce n'est pas tout à fait le cas. Si vous savez que parmi vos cellules il y a plusieurs types cellulaires, mais vous ne savez pas qui est quoi, vous devrez passer par une étape de sélection d'un set de gènes que vous savez plus ou moins spécifiques à tel ou tel type cellulaire. Vous pourrez ensuite procéder à un clustering hiérarchique (et/ou ACP) de vos échantillons pour les classer en fonction de la manière dont elles expriment ces gènes. Si vous vous lancez dans ce type d'analyses sans sélectionner au préalable un set de gènes, il y a peu de chance que vous réussissiez à classer correctement vos cellules (ça sent le vécu, non ? 😉 ).
Cette technique apporte avec elle son lot de complications, néanmoins le single-cell sequencing permet une analyse génomique et transcriptomique à l'échelle d'une seule cellule et permet d'affiner encore et toujours plus nos connaissances. Avec ce procédé, il devient possible d'estimer la variabilité intra-tissulaire, d'étudier des stades embryonnaires précoces, de décortiquer la composition des tumeurs ou même encore de retracer les lignées cellulaires au cours du développement.
Références :
- Single-cell sequencing-based technologies will revolutionize whole-organism science [lien]
- Travaux personnels
Laisser un commentaire